










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkAngiologia e Cirurgia Vascular
versão impressa ISSN 1646-706X
Angiol Cir Vasc v.7 n.3 Lisboa set. 2011
Rastreio de trombofilia hereditária no contexto de trombose venosa profunda
Fernando Mota*, Luciana Ricca Gonçalves**, Armando Mansilha***
* Aluno do 6º Ano de Mestrado Integrado em Medicina, Faculdade de Medicina da Universidade do Porto (med05188@med.up.pt)
** MD, Assistente Hospitalar Graduada de Imunohemoterapia, Hospital de São João
*** MD, PhD, FEBVS, Assistente Hospitalar de Angiologia e Cirurgia Vascular, Professor Convidado da Faculdade de Medicina da Universidade do Porto, Orientador do Projecto de Opção de Mestrado Integrado em Medicina
|RESUMO|
A trombose venosa profunda é uma doença frequente e importante que se manifesta em indivíduos com factores de risco conhecidos ou desconhecidos. A sua etiopatogenia é multifactorial incluindo factores adquiridos e factores genéticos. Dois tipos de defeitos genéticos podem causar trombose venosa: mutações que resultam em deficiência dos inibidores naturais da coagulação e mutações com aumento do nível/função dos factores da coagulação.
O objectivo deste trabalho é referir e discutir as situações em que se deve rastrear a presença de trombofilia hereditária no contexto de um episódio de trombose venosa profunda.
Foram relatadas como factor de risco para trombose venosa, por ordem cronológica, a deficiência de antitrombina, deficiência de proteína C e proteína S, factor V Leiden, mutação G20210A do gene da protrombina e os níveis elevados de factor VIII.
Apesar da associação entre trombofilia hereditária e o risco de trombose venosa estar bem documentada, o mesmo não ocorre em relação ao risco de recorrência. A única situação em que o risco de recorrência foi documentado foi em doentes jovens com deficiência de inibidores naturais da coagulação no contexto de um primeiro episódio de trombose venosa e/ou uma história familiar positiva para trombose venosa.
Actualmente não existe consenso sobre o rastreio de trombofilia hereditária no contexto de trombose venosa profunda. Seria importante que fossem seguidas as guidelines actuais, no sentido de uniformizar a abordagem aos doentes com trombose venosa profunda e facilitar a realização de estudos que permitam elaborar novas guidelines com recomendações baseadas em evidência de elevada qualidade.
Palavras-chave: Trombose venosa profunda, Trombofilia hereditária, Rastreio, Inibidores naturais da coagulação, Factores da coagulação
Screening of familiar trombophylia in patients with deep venous thrombosis
|ABSTRACT|
Deep vein thrombosis is a common and important disease that occurs in individuals with known or unknown risk factors. Its pathogenesis is multifactorial and includes genetic and acquired factors. Two types of genetic defects can cause venous thrombosis: mutations that result in deficiency of natural inhibitors of coagulation and mutations that increase the level / function of coagulation factors.
The aim of this work is to refer and discuss the situations in which the presence of inherited thrombophilia should be screened in the context of an episode of deep vein thrombosis.
The deficiency of antithrombin, protein C and protein S, factor V Leiden, mutation G20210A in prothrombin gene and elevated levels of factor VIII, in chronological order, have been reported as a risk factor for venous thrombosis.
Despite the association between inherited thrombophilia and the risk of venous thrombosis being well documented, the same does not apply for the risk of recurrence. The only situation where the risk of recurrence has been documented is in young patients with deficiency of natural inhibitors of coagulation in the context of a first episode of venous thrombosis and / or a positive family history of venous thrombosis.
Currently there is no consensus on screening for inherited thrombophilia in the context of deep vein thrombosis. It would be important to follow the current guidelines in order to standardize the approach to patients with deep vein thrombosis and facilitate the conduct of studies to elaborate new guidelines with high quality evidence-based recommendations.
Key words: Deep vein thrombosis, Hereditary thrombophilia, Sreening, Natural inhibitors of coagulation, Coagulation factors
LISTA DE ABREVIATURAS E SIGLAS
ACO Anticoncepcionais orais
AD Autossómica dominante
AT Antitrombina
FV Factor V
FVa Factor V activado
FVIII Factor VIII
FIX Factor IX
FVL Factor V Leiden
HC Homocisteína
HHC Hiperhomocisteínemia
MTHFR Metilenotetrahidrofolato Reductase
PC Proteína C
PS Proteína S
RPCa Resistência à Proteína C activada
TEP Tromboembolismo Pulmonar
TEV Tromboembolismo Venoso
TV Trombose Venosa
TVP Trombose Venosa Profunda
INTRODUÇÃO
A trombose venosa (TV) é uma doença frequente com uma incidência anual de 1 a 3 em 1000 por ano[1]. A sua frequência aumenta com a idade, atingindo 1% por ano nos idosos; no entanto esta patologia representa a maior causa de morbilidade e mortalidade durante a gravidez e o parto, e é uma causa frequente de doença em mulheres jovens que tomam contraceptivos orais[1]. Pode ocorrer como trombose venosa profunda (TVP), frequentemente nas veias das pernas (nomeadamente a veia femoral ou a veia poplítea) ou nas veias pélvicas, ou como tromboembolismo pulmonar (TEP).
As principais complicações da TV são uma síndrome pós-trombótica incapacitante (20% dos indivíduos com TV) e morte súbita devido a TEP (1 a 2% dos indivíduos com TV)[2].
A TVP dos membros inferiores é uma situação médica frequente e importante que se manifesta em indivíduos com factores de risco conhecidos ou desconhecidos. Mais de um milhão de casos de TVP são diagnosticados nos E.U.A anualmente, podendo originar até 20000 mortes/ano por TEP.[3]
A etiopatogenia da TV é multifactorial incluindo factores adquiridos assim como factores genéticos. Os factores de risco adquiridos para TV incluem imobilização, cirurgias major e ortopédicas, traumatismo, gravidez, puerpério, síndrome antifosfolipídeo, neoplasia e estrogenoterapia, entre outros[2].
Resultados de vários estudos genéticos estabeleceram que existem dois tipos de defeitos genéticos que podem causar TV: mutações que resultam em deficiência dos inibidores naturais da coagulação e mutações que levam ao aumento do nível/função dos factores da coagulação[1]. O primeiro grupo de mutações aumenta o risco para trombose quando afecta os inibidores naturais da coagulação, nomeadamente a antitrombina (AT), a proteína C (PC) e a proteína S (PS)[4,5,6]. O segundo grupo de mutações afecta o factor V (FV), resultando num FV mutante conhecido como Factor V Leiden (FVL), com aumento da resistência à inactivação feita pela proteína C, e a protrombina (PT), levando a um aumento dos níveis basais de PT (mutação G20210A do gene da PT)[1]. Estas duas mutações são as mais frequentes entre a população caucasiana e têm prevalência quase nula entre a população de raça negra e asiática[7]. As diversas trombofilias hereditárias serão descritas em detalhe adiante.
No entanto, estas alterações muitas vezes não são investigadas ou documentadas. Esta situação pode acontecer devido ao facto de a trombose ser frequentemente resultado de uma combinação de causas, factores de risco e condições predisponentes; assim, um doente portador do factor V Leiden (FVL) pode ser assintomático até ser submetido a uma cirurgia e, posteriormente a um longo período de imobilização, altura em que desenvolve uma TVP que, aparentemente, tem uma causa óbvia, não se prosseguindo a avaliação do doente com um estudo de trombofilia[8].
Para além disso, só recentemente foram identificadas as mutações do FVL e a mutação G20210A do gene da PT – 1993 e 1996, respectivamente[9,10].
Assim, apesar dos avanços no conhecimento sobre os factores de risco hereditários que predispõem à TV, ainda não existe consenso sobre quem deve ser estudado para a presença destas anomalias.
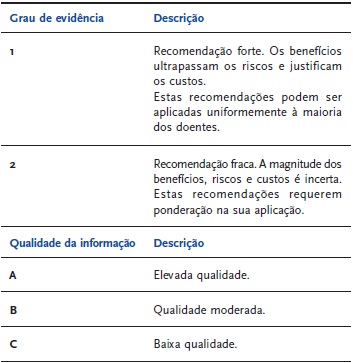
O objectivo deste trabalho é referir e discutir as situações em que se deve rastrear a presença de trombofilia hereditária no contexto de um episódio de TVP. As recomendações e evidências descritas ao longo deste trabalho são baseadas nas guidelines lançadas em Janeiro de 2010 pelo British Committee for Standards in Haematology, tal como sumarizado no | QUADRO 1 |[11].
| QUADRO 1 | Graus de evidência e qualidade da informação[11]
TROMBOFILIA HEREDITÁRIA (DEFINIÇÃO E DESCRIÇÃO)
Trombofilia hereditária é o conjunto de condições genéticas que aumentam o risco de doença tromboembólica e que podem ser causadas por insuficiente inibição da cascata de coagulação, quer por mutações que resultam em deficiência dos inibidores naturais da coagulação, quer por mutações que levam ao aumento do nível/função dos factores da coagulação[1]. O | QUADRO 2 |[12] pretende comparar a prevalência de trombofilia hereditária na população geral e em doentes com tromboembolismo venoso (TEV).
As principais trombofilias hereditárias serão abordadas de seguida.
| QUADRO 2 | Prevalência de trombofilia hereditária[12]
Resistência à proteína C activada e Factor V Leiden
A resistência à proteína C activada (RPCa) é a causa mais frequente de trombofilia hereditária. Resulta, na maior parte das vezes, de uma mutação pontual no gene do FV (mutação R506Q) com substituição da glutamina pela arginina na posição 506 do factor V activado (FVa). O FVa mutante (FV R506Q), comummente designado por FV Leiden (FVL) é resistente à inactivação pela PC activada porque perde um dos locais de acção proteolítica desta enzima. O FVL é o factor de risco para trombose mais prevalente na população caucasiana (3-7%); no entanto é raro nas populações nativas de África ou da Ásia[12]. Nos heterozigóticos representa um risco 3-8 vezes superior ao da população geral para TVP, enquanto os homozigóticos têm um risco cerca de 80 vezes superior[8]. Este risco aumenta significativamente quando estão presentes outros factores de risco como a gravidez, cirurgia, anticoncepcionais orais (ACO) ou outros[13]. Importa referir que a RPCa pode ocorrer, embora raramente, na ausência da mutação do FVL, devido a outros factores genéticos (FV Hong Kong R306G e FV Cambridge R306T) ou mesmo de forma não hereditária, mas associada a factores adquiridos, como por exemplo utilização de ACO[12].
Mutação G20210A do gene da protrombina
Descrita pela primeira vez em 1996, esta mutação consiste na substituição da guanina pela adenina na posição 20210 do gene da PT, numa região não transcrita deste gene[10]. Essa mutação aumenta os níveis de protrombina em circulação e, como a protrombina é um precursor da trombina, ocorrerá um aumento secundário nos níveis de trombina e consequentemente um estado de hipercoagulabilidade. A prevalência desta mutação é de 0,7-4% na população geral e resulta num risco aumentado em cerca de 2-3 vezes para o desenvolvimento de TV [8,12,14,15]. Tal como o FVL, é rara nos indivíduos de raça negra e nos asiáticos.
Deficiência de AT, PC e PS
Já foram descritas inúmeras mutações, de transmissão autossómica dominante (AD), em doentes com défice de PC, PS ou AT.
A AT é um anticoagulante natural que inibe virtualmente todas as proteases da coagulação, mas com maior potência o factor Xa e a trombina (IIa).
Estão definidos dois tipos de deficiência de AT [16,17]: o tipo I, deficiência de AT clássica, é a mais comum e consiste numa deficiência quantitativa com níveis de AT no plasma inferiores a metade do valor normal. Na deficiência de AT tipo II, os níveis plasmáticos de AT estão dentro dos limites da normalidade, mas a actividade da AT está diminuída devido à produção de uma variante do normal. A sua deficiência tem uma prevalência de 0,02% na população geral, e manifesta-se geralmente por TVP dos membros inferiores, EP ou trombose das veias mesentéricas, em doentes com menos de 35 anos e sem outros factores de risco. A deficiência de AT é a trombofilia hereditária mais grave e acarreta um risco relativo para TV de 8,1 e uma incidência anual de trombose de 0,87-1,6% em indivíduos heterozigóticos, a mais elevada entre todas as trombofilias hereditárias[7,8].
A PC actua inactivando o FVa e o factor VIIIa; necessita da PS como cofactor e é activada pela trombina quando esta se liga à trombomodulina endotelial. A PS existe em duas formas: circula livre no plasma, com acção anticoagulante como cofactor da PC ou ligada à proteína de fase aguda C4b-binding protein, não tendo, nesta forma, actividade anticoagulante. No entanto as funções da PS não se limitam à de cofactor da PC, a PS livre também inibe directamente os complexos protrombinase e tenase. A síntese de PS e de PC dá-se no fígado e depende da vitamina K.
Existem dois tipos de deficiência da PC. No tipo I há uma deficiência quantitativa de PC no sangue, sendo esta a forma mais comum de deficiência da PC, resultando de diminuição da síntese ou da estabilidade da PC. No tipo II, a actividade da PC está mais reduzida que os níveis de antigénio o que revela a ocorrência de síntese de moléculas de PC anormais. O gene da PC (PROC) pode sofrer inúmeras mutações (são hoje conhecidas 160) com perda de função que levam ao fenótipo de deficiência de PC[12].
Quanto à deficiência da PS, estão descritos três tipos. No tipo I, aparecem diminuídos os níveis de PS total (deficiência quantitativa). No tipo II, a actividade da PS como cofactor está diminuída, mas existem valores normais de PS total e livre (deficiência qualitativa), sendo um distúrbio muito raro e difícil de diagnosticar. No tipo III estão diminuídos os níveis de PS livre, mas normais os níveis de PS total (deficiência quantitativa de PS livre).
Os défices de PC e de PS têm prevalências de cerca de 0,2-0,4% e de 0,03-0,13%, respectivamente, na população geral[12], e manifestam-se geralmente da mesma forma: TVP dos membros inferiores, TV mesentéricas, TV renais, tromboses dos seios venosos cerebrais ou tromboflebites superficiais, em indivíduos com menos de 25-30 anos[18]. A incidência anual de trombose é de 0,43-0,72% e de 0,5-1,65% para os portadores de défices de PC e PS, respectivamente[7]. Os portadores são quase sempre heterozigóticos. Nos portadores homozigóticos, a deficiência apresenta-se precocemente como purpura fulminans neonatal ou através de TV maciças, e é geralmente fatal. De referir ainda que a diminuição dos níveis de PC e de PS pode ter uma causa adquirida[18].
Polimorfismo C677T do gene da metilenotetrahidrofolato reductase em homozigotia ou heterozigotia e hiperhomocisteínemia
A Homocisteína (HC) é um aminoácido com um grupo sulfidril, derivado da metionina, e é metabolizado através de uma reacção de remetilação ou de trans-sulfuração. As formas adquiridas de hiperhomocisteínemia (HHC) causam elevação leve a moderada dos níveis de HC e podem ser causadas por, entre outras: baixa ingestão de piridoxina, cobalamina e folato. Elas podem produzir HHC ao interagir com factores genéticos como o polimorfismo C677T do gene da metilenotetrahidrofolato reductase (MTHFR), enzima importante no metabolismo da HC, conhecido por variante termolábil (MTHFR C677T), o que leva a uma redução de mais de 50% da actividade da enzima.
A prevalência, na população caucasiana, deste genótipo em heterozigotia é de cerca de 34-37% e em homozigotia de 13,7%[19], sendo semelhante nos indivíduos com eventos tromboembólicos. Assim, a sua associação com um risco aumentado para o desenvolvimento de TV é ainda controversa e não parece muito útil o rastreio deste polimorfismo como factor de risco para eventos tromboembólicos[7,8,20].
Níveis elevados de factores VIII, IX, XI
A presença de níveis basais elevados de factor VIII (FVIII), assim como de outros factores, tem provavelmente uma causa genética, embora ainda não tenha sido identificado um polimorfismo ou mutação em concreto[8], e está associada a um risco aumentado para o desenvolvimento de TV.
Tem uma elevada prevalência na população geral (cerca de 11% tem níveis superiores a 150 UI/dL) sendo que o risco relativo para o desenvolvimento de TV é cerca de 5 vezes superior ao daqueles que têm níveis inferiores a 100UI/dL, e aumenta em 10% por cada 10UI/dL de aumento nos níveis de FVIII[21].
Importa salientar que em alguns laboratórios as baterias de testes para trombofilia apenas incluem os níveis de FVIII e não os dos factores IX e XI (FIX e FXI)[12].
TROMBOFILIA HEREDITÁRIA COMO FACTOR DE RISCO PARA TVP
O impacto de um factor de risco é uma função da sua prevalência e risco relativo. Das várias trombofilias hereditárias descritas, as que foram relatadas como factor de risco para TV foram, por ordem cronológica, a deficiência de AT (4), deficiência de PC[5] e PS[6], FVL, mutação G20210A do gene da PT[1] e os níveis elevados de FVIII[21].
As deficiências de PC, PS e AT são raras, mesmo entre os indivíduos com trombose. Uma vez que estas deficiências são raras, o risco não é facil de avaliar. Foi feita uma estimativa de que estes defeitos aumentam o risco de TVP em, pelo menos, 10 vezes[2].
A RPCa, causada pelo FVL, ocorre em cerca de 5% da população caucasiana[22]. Entre os indivíduos com TV, a mutação do FVL está presente entre 17,6% e 19,5%[22,23] e aumenta o risco de TV em cerca de 8 vezes entre os heterozigóticos[22]. O rastreio para o FVL parece portanto ser pertinente em indivíduos com uma primeira apresentação de TVP.
A mutação G20210A do gene da protrombina tem uma prevalência de cerca de 2,5% na população geral. Entre os doentes com TV, esta mutação foi encontrada em 6% e resulta num risco aumentado em cerca de 2-3 vezes para o desenvolvimento de TV[10]. Esta mutação foi relatada principalmente em pessoas de raça caucasiana[24].
A prevalência de altas concentrações do FVIII depende dos valores de cut-off que são aplicados[2]. Concentrações do FVIII superiores a 150 IU/dL (150% do normal) foram encontradas em 11% da população geral, e em 25% dos doentes com trombose[25]. Tais concentrações estão associadas a um risco de trombose 6 vezes superior quando comparadas com concentrações inferiores a 100 IU/dL[25].
TROMBOFILIA HEREDITÁRIA E O RISCO DE TV RECORRENTE
Nas décadas de 1980 e 1990 os testes para trombofilia hereditária tornaram-se comuns em doentes não seleccionados e nos seus familiares, apesar do facto de não existir evidência da sua utilidade clínica[11]. A evolução sobre o conhecimento dos factores protrombóticos e a sua associação com o primeiro evento trombótico despoletou o entusiasmo no diagnóstico de trombofilias, especialmente com o objectivo de definir o risco individual de recorrência de TV.
Contudo, apesar de a associação entre factores protrombóticos e o risco de TV estar bem documentada, o mesmo não ocorre em relação ao risco de recorrência[26,27,28,29].
Alguns estudos demonstraram que o rastreio para trombofilia hereditária não possui valor preditivo quanto à recorrência de um evento trombótico em doentes não seleccionados com TV sintomática[26,27], assim como não reduz a recorrência de TV[28].
Num estudo de corte prospectivo, verificou-se que o risco de recorrência de TVP em pacientes jovens não está relacionado com a presença ou ausência de evidência laboratorial da presença de trombofilia hereditária[29]. Nesse estudo, o risco de recorrência em indivíduos com a mutação do FVL foi de 1,26 e em indivíduos com a mutação G20210A do gene da protrombina foi de 0,81[29].
Num outro estudo, o risco de recorrência após um primeiro episódio de trombose venosa foi avaliado prospectivamente em 474 indivíduos com idade inferior a 70 anos, sem doença oncológica (follow-up de 7,3 anos)[27]. Neste estudo, globalmente, os factores pró-trombóticos considerados (FVL, mutação da PT, deficiência em inibidores naturais da coagulação, FVIII, FIX, FXI e HHC) não modificaram o risco de recorrência. A deficiência de inibidores naturais da coagulação (AT, PC e PS) e a presença de outras trombofilias combinadas induziram um aumento ligeiro do risco de recorrência (risco relativo 1,8 e 1,6, respectivamente).
As trombofilias hereditárias mais frequentemente detectadas, o FVL na forma heterozigótica e a mutação G20210A da PT na forma heterozigótica, têm um efeito relativamente pequeno sobre o risco de recorrência. Revisões sistemáticas sobre o risco de recorrência de TV em doentes com esses defeitos demonstraram um risco de 1,4 para a mutação do FVL e de 1,2-1,7 para a mutação G20210A da PT[30,31]. Os autores concluíram que o aumento do risco era modesto e que, por si só, não justificaria um prolongamento da terapêutica anticoagulante.
Alguns estudos realizados contemplando as restantes trombofilias hereditárias também demonstraram um efeito modesto sobre o risco de recorrência; em doentes não seleccionados com deficiência das proteínas anticoagulantes (AT, PC e PS) o risco relativo de recorrência foi inferior a 2,0[26,27].
Por outro lado, os doentes com história familiar de trombofilia e TV são considerados como tendo um risco de recorrência superior[32,33]. Pensa-se que na base desse maior risco esteja uma eventual transmissão hereditária de outros factores trombofílicos não mensuráveis[32,33]. De facto, é consensual que o risco de recorrência é superior em doentes que sejam portadores de mais de uma trombofilia em comparação aos portadores de apenas uma[32,33].
Um estudo retrospectivo de um grupo de doentes com idade jovem na altura da primeira TV e com história familiar de TV concluiu que a detecção de uma deficiência num inibidor natural da coagulação (AT, PC, PS) comporta uma taxa de recorrência anual de 6,23%, comparado com uma taxa de 2,25% em doentes com as mutações do FVL, mutação G20210A da PT ou níveis elevados de FVIII[34]. Após um período de 10 anos, a taxa de recorrência cumulativa atingia os 55% nos doentes com deficiências de AT, PC ou PS enquanto para os doentes com a mutação do FVL, a mutação G20210A da PT ou níveis elevados de FVIII essa taxa era de 25%[34]. Os autores concluem que, dadas as suas implicações clínicas, o rastreio de trombofilia hereditária deveria ser realizado com o intuito de detectar deficiência de AT, PC ou PS em doentes jovens com um primeiro episódio de TV e/ou uma história familiar positiva para trombose venosa[34].
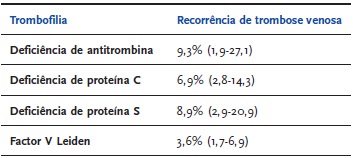
Um outro estudo prospectivo observacional, realizado em doentes com história familiar de trombofilia, alertou para o maior risco de recorrência em doentes do sexo masculino portadores de deficiências de inibidores naturais da coagulação ou de trombofilias combinadas[35]. Nos indivíduos portadores de deficiência de AT, a taxa de recorrência anual foi mais elevada (9,3%), ocorrendo mesmo naqueles sob terapêutica anticoagulante de longa duração (2,7%)[35]. O | QUADRO 3 |[35]pretende comparar a taxa anual de recorrência de TV em doentes com diferentes trombofilias hereditárias. Os autores salientaram o facto de a recorrência ocorrer em contextos de risco elevado e, por isso, potencialmente evitáveis por uma estratégia adequada de prevenção secundária[35].
| QUADRO 3 | Taxa anual de recorrência de trombose venosa[35]
RASTREIO DE TROMBOFILIA HEREDITÁRIA E TRATAMENTO DE TVP
Como já foi referido, apesar da associação entre factores protrombóticos e o risco de TV estar bem documentada, o mesmo não ocorre em relação ao risco de recorrência. A única situação em que o risco de recorrência foi documentado foi em doentes jovens com deficiência de AT, PC ou PS no contexto de um primeiro episódio de TV e/ou uma história familiar positiva para trombose venosa[34].
Num artigo de revisão em que foram analisados 70 eventos trombóticos em 57 indivíduos com deficiência de AT, a recorrência e extensão da trombose durante o tratamento não se revelaram superiores ao observado em doentes tratados para TV em que não tenha sido detectada essa deficiência[36].
A intensidade do tratamento de manutenção com anticoagulantes orais não deve ser alterada por evidência laboratorial de trombofilia hereditária [11]. Não existe evidência de que a recorrência de trombose após instituição de tratamento com anticoagulante oral seja superior em doentes com trombofilia hereditária comparativamente aos que não têm trombofilia[37].
As guidelines lançadas em Janeiro de 2010 pelo British Committee for Standards in Haematology[11] fazem as seguintes recomendações em relação a este tópico:
> Após um diagnóstico de TV aguda, o início e a intensidade da terapêutica anticoagulante devem ser os mesmos em doentes com ou sem trombofilia hereditária (grau de evidência 1B)[11];
> Testar indiscriminadamente a presença de trombofilia hereditária em doentes não seleccionados que se apresentem com um primeiro episódio de TV não está indicado (grau de evidência 1B)[11];
> As decisões referentes à duração da anticoagulação (vitalícia ou não) em doentes não seleccionados devem ser tomadas tendo em conta se um primeiro episódio de TV foi provocado ou não, outros factores de risco, e o risco de hemorragia relacionada com a terapêutica anticoagulante, independentemente de existir ou não uma trombofilia hereditária (grau de evidência 1B)[11];
> Testar a presença de trombofilia hereditária em doentes seleccionados, como aqueles com história familiar de TV recorrente não provocada, pode influenciar decisões referentes à duração da terapia anticoagulante (grau de evidência C). No entanto, não é possível dar uma recomendação validada em relação a como esses doentes devem ser seleccionados[11].
RASTREIO DE TOMBOFILIA HEREDITÁRIA EM FAMILIARES DE DOENTES COM TVP
É importante perceber se o rastreio de trombofilia hereditária em familiares assintomáticos de doentes com TVP tem, ou não, utilidade clínica. Utilidade clínica é a probabilidade de um teste permitir um melhor resultado em termos de saúde para o doente[38]. No caso do rastreio de trombofilia hereditária temos de questionar se é provável que esse rastreio conduza a um melhor resultado em termos de saúde e se vale a pena os custos, não só financeiros mas também psicossociais[38].
Alguns argumentos a favor do rastreio são que ele pode permitir a evicção de riscos adquiridos (como o uso de contraceptivos orais nas mulheres) e a optimização da estratégia profiláctica em situações de alto risco (como cirurgia, imobilização prolongada no leito por doença, viagens longas, etc.). Este conhecimento poderia assim permitir elaborar uma estratégia com vista à prevenção primária de trombose venosa, e contribuir para reduzir a incidência de trombose venosa nas famílias e, consequentemente, na população em geral[32].
No caso específico da mulher, a identificação de trombofilia tem implicações na selecção do método de planeamento familiar, já que contra-indica a contracepção oral estroprogestativa (factor de risco independente de trombose venosa)[32]. Do mesmo modo, o tratamento hormonal de substituição (oral) está contra-indicado[32].
No entanto, o risco individual é afectado por múltiplos factores genéticos e ambientais, que variam mesmo entre parentes de primeiro grau[11].
Esta questão foi abordada objectivamente por uma série de estudos.
Um estudo prospectivo avaliou a incidência de TV em 470 portadores assintomáticos da mutação do FVL[39]. Os portadores foram identificados através do rastreio de parentes em primeiro grau de 247 doentes sintomáticos. A incidência anual total de TV foi de 0,58% com uma incidência absoluta de episódios espontâneos de apenas 0,26%[39]. Os autores concluíram que a incidência anual absoluta de TV espontânea em portadores assintomáticos da mutação do FVL é baixa e que não justifica o rastreio de rotina nas famílias de doentes sintomáticos[39].
Um estudo prospectivo semelhante verificou que o risco anual total de TV em 313 portadores assintomáticos da mutação do FVL, aparentados de 131 doentes sintomáticos, foi de 0,67%. Com um risco de TV espontânea de apenas 0,17%[40].
O valor preditivo parece ser maior para a deficiência de um inibidor natural da coagulação. Um estudo de coorte prospectivo verificou que a incidência anual de TV em portadores assintomáticos de deficiência de AT, PC ou PS foi de 1,5%, tendo cerca de metade dos episódios sido provocados[41].
Um estudo retrospectivo avaliou o risco de TV em familiares assintomáticos de doentes com trombofilia hereditária e uma história familiar ou pessoal de TV[42]. O risco de TV foi 16 vezes superior nos familiares com trombofilia, tendo o risco mais alto ocorrido nos familiares de doentes com uma deficiência de um anticoagulante natural ou com defeitos múltiplos[42]. Foi realizado posteriormente um estudo prospectivo a esses doentes, com um follow-up de cerca de 6 anos, onde se verificou que 4,5% de 575 portadores assintomáticos de trombofilia sofreram um primeiro episódio de TV, comparativamente a 0,6% de um grupo controlo. Cerca de 60% dos episódios foram espontâneos[43]. A incidência de TV foi de 0,8% por ano nos portadores e de 0,1% por ano nos controlos. A incidência mais alta ocorreu nos portadores de deficiência de AT (1,7% por ano) ou nos com defeitos combinados (1,6% por ano)[43].
Actualmente ainda não foi demonstrada uma relação custo-eficácia para o rastreio de trombofilia hereditária em famílias com historial de trombose. Os métodos simples para determinação de uma história familiar positiva não discriminam a presença ou ausência de trombofilia e portanto a decisão de realizar rastreio não pode ser baseada somente na existência ou inexistência de uma história familiar[11].
As guidelines lançadas pelo British Committee for Standards in Haematology[11]fazem as seguintes recomendações em relação a este tópico:
O rastreio de familiares assintomáticos para a presença de trombofilia hereditária de baixo risco, tal como a mutação do FVL ou a mutação G20210A da PT, não está indicado (grau de evidência 1B)[11];
O rastreio de familiares assintomáticos para a presença de trombofilia hereditária de alto risco, tal como a deficiência de AT, PC ou PS, só deve ser considerado em famílias com tendência trombótica (trombosis-prone families) (grau de evidência 1B). Se o rastreio for efectuado, os riscos, os benefícios e as limitações do rastreio devem ser discutidas juntamente com uma explicação da hereditariedade e do risco da doença. No entanto, não é possível dar uma recomendação em relação a como esses doentes e famílias devem ser seleccionados[11];
O rastreio para a presença de trombofilias homozigóticas ou compostas raras não está indicado pois estes defeitos são tão raros que não podem ser previstos por história familiar, e o risco de trombose não provocada é baixo (grau de evidência 2C)[11].
TIMING DO RASTREIO E METODOLOGIA LABORATORIAL
Na fase aguda da trombose venosa e nos indivíduos sob anticoagulação oral alguns resultados laboratoriais podem ser falseados ou difíceis de interpretar.
Idealmente, o rastreio laboratorial de trombofilia deve ser efectuado fora da fase aguda da trombose e no mínimo um mês após suspensão da anticoagulação oral[45]. Por exemplo, a PC e PS, sendo dependentes da vitamina K, encontram-se frequentemente diminuídas nos indivíduos sob terapêutica anticoagulante oral[44].
É também desejável um intervalo mínimo de um mês entre o doseamento da PS e a suspensão de tratamento hormonal ou o fim da gravidez[45]. A gravidez e as terapêuticas com estrogénios (contracepção oral ou terapêutica hormonal de substituição) diminuem a PS circulante[44].
O doseamento de AT na fase aguda da trombose venosa constitui uma excepção e pode, em certos casos, ser importante. Quando há défice de AT, pode, por vezes, haver indicação para a administração de concentrados de AT em associação à heparina não fraccionada ou à heparina de baixo peso molecular[45].
A presença de défice de AT, PC e PS deve ser confirmada em várias ocasiões (mínimo dois doseamentos, na ausência de interferência por factores externos)[11].
Os testes genéticos podem ser realizados em qualquer altura, uma vez que os seus resultados não são influenciados por factores externos.
As recomendações das guidelines lançadas pelo British Committee for Standards in Haematology[11] quanto à realização dos testes laboratoriais para identificação de trombofilia hereditária e sua interpretação são as seguintes:
A realização dos testes laboratoriais durante um episódio agudo de TV não está indicada pois a utilidade e implicações desses testes devem ser antes consideradas e o doente deve ser consultado antes da sua realização. Uma vez que o tratamento da TV aguda não é influenciado pelos resultados dos testes, estes podem ser efectuados posteriormente, se indicado[11];
O tempo de protrombina deve ser medido para detectar o efeito dos anticoagulantes orais dependentes da vitamina K, que causam uma redução nos níveis de PC e PS[11];
Estudos funcionais devem ser utilizados para determinar os níveis de AT e PC[11];
Estudos cromogénicos para determinar a actividade da PC estão menos sujeitos a interferência do que os estudos de coagulação, e são preferíveis[11];
Estudos imunorreactivos para quantificação do antigénio da PS livre são preferíveis a estudos funcionais. Se for realizado um estudo da actividade da PS como parte de um rastreio inicial, os resultados baixos devem ser posteriormente investigados recorrendo a um estudo imunorreactivo da PS livre[11];
Se for realizado um estudo de RPCa para detectar a mutação do FVL, então deve ser efectuado o teste modificado de sensibilidade à PC activada (pré-diluição da amostra teste em plasma deficiente em FV) por oposição ao teste original de sensibilidade à PC activada. Se o teste for positivo a presença da mutação deve ser confirmada por teste genético directo. A realização de um estudo de RPCa não é necessária se um teste genético directo para a mutação do FVL for realizado inicialmente[11];
Os testes para identificação de deficiência de AT, PC e PS devem ser repetidos e os níveis baixos devem ser confirmados em uma ou mais amostras separadas. Um único resultado anormal não é suficiente para fazer diagnóstico destas deficiências[11];
Os testes para trombofilia hereditária devem ser supervisionados por profissionais experientes, e os resultados bem como as suas implicações clínicas devem ser interpretados por um clínico experiente que esteja ciente de todos os factores que podem influenciar os resultados dos testes em cada caso específico[11].
Os laboratórios que executam testes de trombofilia devem estar sujeitos a controlos laboratoriais internos e externos rigorosos e acreditados por entidades oficiais.
CONCLUSÃO
Actualmente ainda não existe consenso sobre o rastreio de trombofilia hereditária no contexto de TVP. Se olharmos para as guidelines lançadas em Janeiro de 2010 pelo British Committee for Standards in Haematology, podemos verificar que nenhuma das recomendações feitas a este propósito possui grau de evidência A, o que significa que essas recomendações não estão consistentemente corroboradas por ensaios clínicos controlados e randomizados. Assim, é natural que as opiniões continuem a divergir e que ainda haja espaço para a investigação e para a obtenção de dados mais definitivos.
No entanto, se aplicadas, as guidelines actuais podem trazer benefícios não só para o doente mas também para os seus familiares e os próprios profissionais de saúde. Podem diminuir o número de testes para a detecção de trombofilia hereditária realizados sem necessidade e que não apresentam utilidade clínica, reduzindo assim os gastos dos serviços de saúde; podem melhorar a abordagem ao doente após ser detectada uma trombofilia hereditária consoante o tipo de trombofilia e assim optimizar o tratamento destes doentes; podem ajudar à decisão de quando rastrear os familiares de um doente com uma trombofilia hereditária e permitir um melhor aconselhamento do doente e dos familiares quanto à sua doença (evicção de outros factores de risco para TVP, por exemplo).
A interpretação dos resultados dos testes laboratoriais para trombofilia hereditária é difícil porque:
A incidência de trombose em indivíduos portadores de trombofilia hereditária é muito variável;
Muitos indivíduos com resultado laboratorial positivo nunca tiveram um evento trombótico;
A ausência de identificação laboratorial não significa ausência de trombofilia;
Os clínicos podem sobrestimar a existência de trombofilia e menosprezar os riscos da anticoagulação.
Parece consensual, perante os dados actuais, que o rastreio para as trombofilias mais frequentes (aumento do nível/função dos factores da coagulação) não terá utilidade clínica, pois não aumentam o risco de recorrência de TVP e o tratamento não é influenciado pela sua detecção.
Mais controverso é o rastreio para trombofilias hereditárias raras (deficiência de inibidores naturais da coagulação) que aumentam o risco de recorrência e podem ter implicações na terapêutica. Serão necessários mais estudos para definir claramente quais os doentes que devem ser submetidos a rastreio para estas trombofilias hereditárias e quais as mudanças que a sua presença implicará na terapêutica desses doentes.
É também necessário encontrar uma definição clara das famílias que terão tendência aumentada para TVP e em que situações os integrantes dessas famílias deverão ser submetidos a rastreio para trombofilia hereditária, nomeadamente para a presença de mutações que afectem os inibidores naturais da coagulação, pois parecem ser estas que justificam essa abordagem.
Serão, portanto, necessários estudos futuros com rigor metodológico que permitam elaborar novas guidelines com recomendações baseadas em evidência de elevada qualidade e que melhorem a abordagem aos doentes com TVP.
BIBLIOGRAFIA
[1] Reitsma PH, Rosendaal FR. Past and future of genetic research in thrombosis. Journal of Thrombosis and Haemostasis 2007;5:264–269. [ Links ]
[2] Rosendaal FR. Venous thrombosis: a multicausal disease. Lancet 1993;353:1167–73. [ Links ]
[3] Kreidy R, Irani-Hakime N. Is thrombophilia a major risk factor for deep vein thrombosis of the lower extremities among Lebanese patients? Vascular Health and Risk Management 2009;5:627–633. [ Links ]
[4] Egeberg O. Inherited Antithrombin Deficiency Causing Thrombophilia. Thrombosis Diathesis Haemorrhagica 1965;13:516–530. [ Links ]
[5] Griffin JH, Evatt B, Zimmerman TS, Kleiss AJ, Wideman C. Deficiency of protein C in congenital thrombotic disease. Journal of Clinical Investigation 1981;68:1370–1373. [ Links ]
[6] Comp PC, Nixon RR, Cooper MR, Esmon CT. Familial protein S deficiency is associated with recurrent thrombosis. Journal of Clinical Investigation 1984;74:2082–2088. [ Links ]
[7] Seligsohn U, Lubetsky A. Genetic susceptibility to venous thrombosis. N Engl J Med 2001;344:1222-1231. [ Links ]
[8] Thomas RH. Hypercoagulability syndromes. Arch Intern Med 2001;161:2433-2439. [ Links ]
[9] Bertina RM, Koeleman BP, Koster T, Rosendaal FR, Dirven RJ, Ronde H, van der Velden, PA, Reitsma PH. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature 1995;369:64–67. [ Links ]
[10] Poort SR, Rosendaal FR, Bertina RM, Reitsma PH. A common genetic variation in the 3-untranslated region of the prothrombin gene is associated with elavated plasma prothrombin levels and increase in venous thrombosis. Blood 1996;88:3698-3703. [ Links ]
[11] British Committee for Standards in Haematology. Clinical guidelines for testing for heritable thrombophilia, British Journal of Haematology 2010;149:209–220. [ Links ]
[12] Middeldorp S, van Hylckama Vlieg A. Does thrombophilia testing help in the clinical management of patients? Br J Haematol 2008;143:321-35. [ Links ]
[13] Vandenbrouke JP, Koster T, et al. Increased risk of venous thrombosis in oral-contraceptive users who are carriers of factor V Leiden mutation. Lancet 1994;344:1453-1457. [ Links ]
[14] Kim V, Spandorfer J. Epidemiology of venous thromboembolic disease. Med Clin North Am 2001;19:839–859. [ Links ]
[15] Mansilha A, Araújo F, Sampaio SM, Cunha Ribeiro LM, Braga A. The PORtromb Project: prothrombin G20210A mutation and venous thromboembolism in young people. Cardiovascular Surgery 2002;10:45-48. [ Links ]
[16] Lane D, et al. Antithrombin III mutation database: first update. For the thrombin and its inhibitors subcommittee of scientific and standardization committee of the International Society on Thrombosis and Haemostasis. Thromb Haemost, 1993;70:361-369. [ Links ]
[17] Lane D, et al. Antithrombin III mutation database: second update. For the plasma Coagulation Inhibitors Subcommittee of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. Thromb Haemost 1997;77:197-211. [ Links ]
[18] Silver D, Vouyouka A. The caput medusae of hypercoagulability. J Vasc Surg 2000;31:396-405. [ Links ]
[19] Varga E. Inherited thrombophilia: key points for genetic counseling. J Genet Couns 2007;16:261-77. [ Links ]
[20] Gemmati D, Serino ML, et al. C677T substitution in the methylenetetrahydrofolate reductase gene as a risk factor for venous thrombosis and arterial disease in selected patients. Haematologica 1999;84:824-8. [ Links ]
[21] Kraaijenhagen R, Pieternella S, Koopman M, et al. High plasma concentrations of factor VIII:C is a major risk factor for venous thromboembolism. Thromb Haemost 2000; 83:5-9. [ Links ]
[22] Rosendaal FR, Koster T, Vandenbroucke JP, Reitsma PH. High risk of thrombosis in patients homozygous for factor V Leiden (activated protein C resistance). Blood 1995;85:1504–08. [ Links ]
[23] Bertina RM, Koeleman BPC, Koster T, et al. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature 1994;369:64-67. [ Links ]
[24] Rosendaal FR, Doggen CJM, Zivelin A, et al. Geographic distribution of the 20210 G to A prothrombin variant. Thromb Haemost 1998;79:706–08. [ Links ]
[25] Koster T, Blann AD, Briët E, Vandenbrouke JP, Rosendaal FR. Role of clotting factor VIII in effect of von Willebrand factor on occurrence of deep-vein thrombosis. Lancet 1995;345:152–55. [ Links ]
[26] Baglin T, et al. Incidence of recurrent venous thromboembolism in relation to clinical and thrombophilic risk factors:prospective cohort study. Lancet 2003;362:523-526. [ Links ]
[27] Christiansen SC, Cannegieter SC, Koster T, Vandenbroucke JP, Rosendaal FR. Thrombophilia, clinical factors and recurrent venous thrombotic events. Journal of the American Medical Association 2005;293:2352–2361. [ Links ]
[28] Coppens M, Reijnders JH, Middeldorp S, Doggen CJ, Rosendaal FR. Testing for inherited thrombophilia does not reduce recurrence of venous thrombosis. Journal of Thrombosis and Haemostasis 2008;6:1474–1477. [ Links ]
[29] Mansilha A, Araújo F, Severo M, Sampaio SM, Toledo T, Albuquerque R. Genetic Polymorphisms and Risk of Recurrent Deep Venous Thrombosis in Young People: Prospective Cohort Study. Eur J Vasc Endovasc Surg 2005;30:545-549. [ Links ]
[30] Ho WK, Hankey GJ, Quinlan DJ, Eikelboom JW. Risk of recurrent venous thromboembolism in patients with common thrombophilia: a systematic review. Archives of Internal Medicine 2006;166:729–736.
[31] Marchiori A, Mosena L, Prins MH, Prandoni P. The risk of recurrent venous thromboembolism among heterozygous carriers of factor V Leiden or prothrombin G20210A mutation. A systematic review of prospective studies. Haematologica 2007;92:1107–1114. [ Links ]
[32] Cushman M. Inherited risk factors for venous thrombosis. Hematology 2005;452-457. [ Links ]
[33] Khan S, et al. Hereditary thrombophilia. Thrombosis Journal 2006;4:15. [ Links ]
[34] Lijfering WM, Brouwer JL, Veeger NJ, Bank I, Coppens M, Middeldorp S, Hamulyak K, Prins MH, Buller HR, van der Meer J. Selective testing for thrombophilia in patients with first venous thrombosis. Results from a retrospective family cohort study on absolute thrombotic risk for currently known thrombophilic defects in 2479 relatives. Blood 2009;113:5314–5322. [ Links ]
[35] Vossen CY, et al. Recurrence rate after a first venous thrombosis in patients with familial thrombophilia. Arterioscl Thromb Vasc Biol 2005;25:1992-1997. [ Links ]
[36] Schulman S, Tengborn L. Treatment of venous thromboembolism in patients with congenital deficiency of antithrombin III. Thrombosis and Haemostasis 1992;68:634–636. [ Links ]
[37] Kearon C, Julian JA, Kovacs MJ, Anderson DR, Wells P, MackinnonmB, Weitz JI, Crowther MA, Dolan S, Turpie AG, Geerts W, Solymoss S, van Nguyen P, Demers C, Kahn SR, Kassis J, Rodger M, Hambleton J, Gent M, Ginsberg JS. Influence of thrombophilia on risk of recurrent venous thromboembolism while on warfarin: Results from a randomized trial. Blood 2008;112:4432–4436. [ Links ]
[38] Baglin T. Management of Thrombophilia: Who to Screen? Pathophysiol Haemost Thromb 2003/2004;33:401-404. [ Links ]
[39] Middeldorp S, Meinardi JR, Koopman MM, et al. A prospective study of asymptomatic carriers of the factor V Leiden mutation to determine the incidence of venous thromboembolism. Ann Intern Med 2001;135:322-327. [ Links ]
[40] Simioni P, Tormene D, Prandoni P, et al. Incidence of venous thromboembolism in asymptomatic family members who are carriers of factor V Leiden: a prospective cohort study. Blood 2002;99:1938-1942. [ Links ]
[41] Sanson B-J, Simioni P, Tormene D, et al. The incidence of venous thromboembolism in asymptomatic carriers of a deficiency of antithrombin, protein C or protein S: a prospective cohort study. Blood 1999;94:3702-3706. [ Links ]
[42] Vossen CY, Conard J, Fontcuberta J, Makris M, Van Der Meer FJ, Pabinger I, Palareti, G, Preston FE, Scharrer I, Souto JC, Svensson P, Walker ID, Rosendaal, FR. Familial thrombophilia and lifetime risk of venous thrombosis. Journal of Thrombosis and Haemostasis 2004;2:1526–1532. [ Links ]
[43] Vossen CY, Conard J, Fontcuberta J, Makris M, Van Der Meer FJ, Pabinger I, Palareti G, Preston FE, Scharrer I, Souto JC, Svensson P, Walker ID, Rosendaal FR. Risk of a first venous thrombotic event in carriers of a familial thrombophilic defect. The European Prospective Cohort on Thrombophilia (EPCOT). Journal of Thrombosis and Haemostasis 2005;3:459–464. [ Links ]
[44] Fonseca AG, Amaro M. Thrombophilias: the importance of clinical screening in thromboembolic disease. Medicina Interna 2008;15:284-290. [ Links ]
[45] Gouault-Heilmann et al. Recomendations pour une juxte prescription des examens dhemostase en pratique médicale courante. STV 2006;18:29-42. [ Links ]

