











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
CLINICAL CASE
We report the case of a four-year-old male, who presented with a one-year history of asymptomatic facial lesions. Weeks later, these lesions progressed a few lesions similar lesions occurred on the proximal upper and lower extremities. The patient was healthy and had no associated extracutaneous symptoms.
On physical examination, we observed several brown flat-topped papules on the face, favouring the malar and zygomatic areas bilaterally, and on the proximal upper and lower extremities (Fig.s 1 and 2). Darier sign was negative.
The patient had no other cutaneous or mucosal lesions. Lesional biopsy was performed, revealing a mild infiltrate in the papillary and superficial portion of the reticular dermis, consisting predominantly of large oval cells (Fig. 3). These cells were slightly pleomorphic and presented abundant pale cytoplasm, with poorly defined limits. Scattered lymphocytes and occasional eosinophils were also present.
The immunohistochemical study revealed a predominance of histiocytic cells in the infiltrate, with strong diffuse positivity for CD163. Only a few scattered cells were positive for S100 protein, CD1a, Langerin and CD117 (c-kit) (Fig. 4).

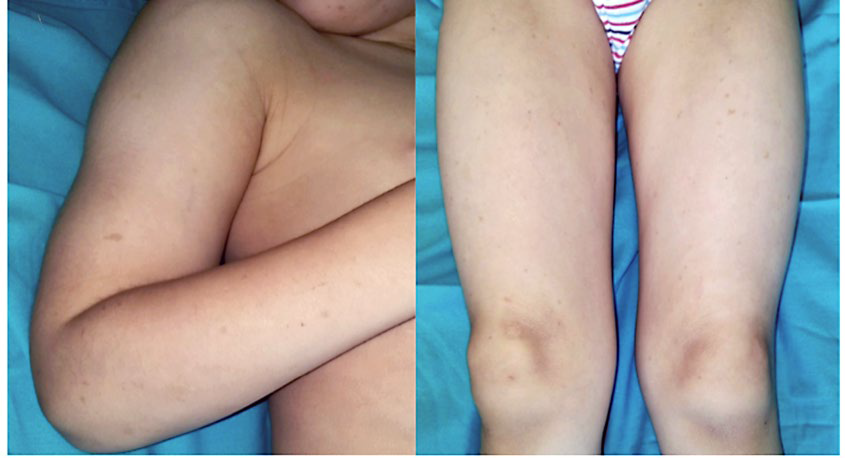

Figure 3 Histopathological findings. A mild infiltrate in the papillary and in the superficial portion of the reticular dermis consisting predominantly of relatively large oval cells. These cells are slightly pleomo rphic and presented abundant pale cytoplasm, with poorly defined limits.

Figure 4 The immunohistochemical study revealed a predominance of histiocytic cells in the infiltrate, with strong diffuse positivity fo r CD163.
BENIGN CEPHALIC HISTYOCITOSIS
These histopathological findings favour a non-Langerhans cell histiocytosis. Taking into account the clinical presentation, namely the patient’s age and the topographic distribution of the lesions, this case can be interpreted as a benign cephalic histiocytosis.
Benign cephalic histiocytosis (BCH) is a rare histiocytic proliferative disorder occurring in young children, primarily affecting the face. It usually presents by the age of one year and is more common in males.1 Systemic involvement in the form of diabetes insipidus has been reported, but is uncommon.2 No treatment is usually needed, as it is generally a self-limiting disorder.
The pathogenesis of BCH is not fully understood. It is intriguing to notice that juvenile xanthogranuloma and generalized eruptive histiocytoma share similar histopathology, ultrastructural appearance, and immunohistochemical profile with BCH (3). In light of these findings, several authors have suggested that these entities should be viewed as a single disease with a spectrum of clinical features.
Regarding histopathological features, there are 3 different histologic patterns described: a papillary dermal pattern, a diffuse pattern, and a lichenoid pattern.3 Our case conforms to the papillary dermal pattern. Histiocytes are often admixed with lymphocytes and a few eosinophils and cells typically express non-Langerhans cell histiocytic markers including CD11b, CD11c, CD14b, CD68 and HAM56.4,5
The differential diagnosis includes the non-Langerhans cell histiocytosis (non-LCH) disorders, particularly eruptive histiocytoma and juvenile xanthogranuloma, as well as urticaria pigmentosa. Frequently, clinical differentiation is possible, as the number of the lesions as well their distribution pattern mainly in the cephalic region usually allow for distinction of benign cephalic histiocytosis from other non-LCH disorders. Immunohistochemical findings are vital for excluding other entities, namely indeterminate cell histiocytosis and Langerhans cell histiocytosis (LCH).
Even though no treatment is usually needed given the benign and self-limiting nature of BCH, regular examinations of all patients are recommended, since periodic exacerbations of the disease and diabetes insipidus are
possible.
Regarding the clinical course, the patient presented with stable lesions, without progression of the disease.
Given the benign nature of the disease, no additional investigation was performed but he remains under observation, twice annually. To this moment, he has a follow-up time of 18 months.

