











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
INTRODUCTION
Cutaneous plasmacytosis is a rare dermatosis that in its classical form is characterized by the presence of multiple reddishbrown papules and nodules, mainly on the trunk. It most often affects elderly Asians,1 with few reports in Caucasians and young individuals.2,3Cutaneous histopathology shows dermal polyclonal proliferation of mature plasma cells, without atypia. Definitive diagnosis tends to be delayed.1
The purpose of this report is to present a case with atypical manifestation of this rare disease, both for clinical and epidemiological aspects.
CASE REPORT
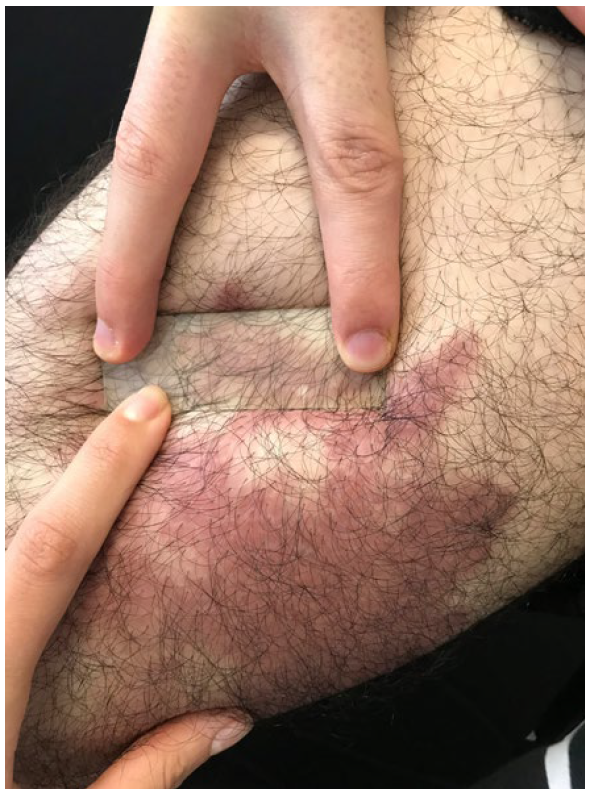
A 21 years-old male patient referred an asymptomatic patch on his right thigh, that began when he was 12 years old, slowly enlarged with a downward progression to the leg and new similar lesions on the contralateral lower limb (Fig.s 1 and 2). On examination, erythematous-purple well demarcated patches on the legs with a geographical pattern or a more reticular pattern on the thighs were partially reducible with diascopy (Fig. 3). Lesions showed no skin indurating and distribution of body hair and sensitivity was preserved. He denied fever and other systemic symptoms. There were no palpable lymph nodes, hepato or splenomegaly on physical examination.
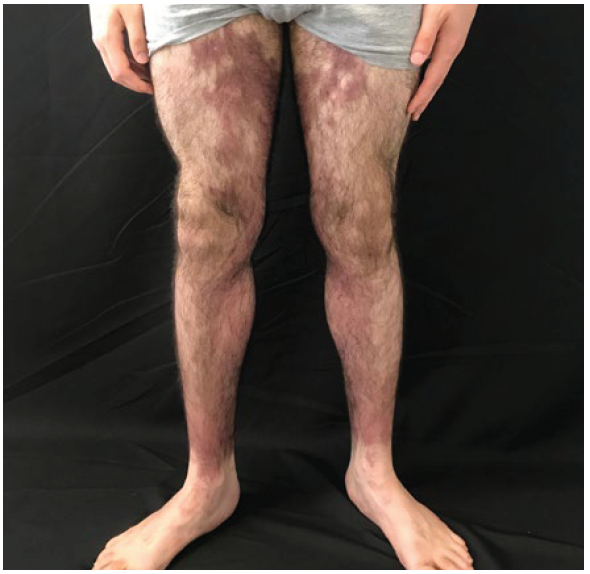
Figure 1 Anterior view of lower limbs: red-brown macules mainly in the medial portion of the thighs and legs, with well-defined limits and a geographical pattern.
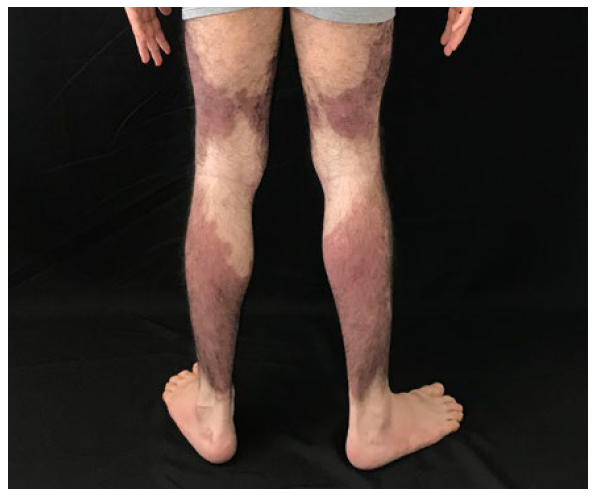
Figure 2 Posterior view of lower limbs: red-brown macules, bilaterally, with a descending linear aspect, with marked symmetry.
Blood cell count and serum biochemistry was normal. Serum pro-tein electrophoresis demonstrated 6.8 g/L of total proteins, with an albumin of 4.37 g/L and an albumin/globulin ratio of 1.80, without a monoclonal peak. Bence-Jones protein urine was negative as well as syphilis and borreliosis serology. Three cutaneous histopathology exams performed on the thighs with 6 years interval, all exhibited the same pattern, however, with a progressive increase in the intensity and representativeness of the findings. In the dermis, throughout the hole thickness of the sample, we observed epithelioid granulomas and perivascular and periadnexal cuffs made up of numerous plasma cells, histiocytes and lymphocytes extending to the interstitial dermis and permeating collagen bundles. Plasma cells were slightly enlarged but without atypia. Central nervous system magnetic resonance imaging and chest, abdomen and pelvis computed tomography scan were normal. No bone marrow investigation was recommended by the Hematologist who evaluated the patient.
There was no improvement with oral colchicine (1 g/day, p.o., for 6 months), doxycycline (200 mg/day, p.o., for 6 weeks) and systemic corticosteroids (prednisolone 1 mg/kg/day during 1 month, with slow dosage tapering and complete withdrawal after 2 months) associated either with mometasone 0.1% cream or clobetasol propionate cream.
After reviewing the last cutaneous histopathology we could cha-racterize a dense superficial and deep perivascular and periadnexal infiltrate composed by numerous plasma cells, histiocytes and lymphocytes permeating collagen bundles with a “rosette” aspect, along with hemosiderin deposits (Fig. 4). Infiltrating cutaneous plasma cells were polyclonal exhibiting both κ- and λ-chains in immunohistochemistry (Fig. 5). Those findings, in correlation with the clinical aspects, suggest the diagnostic hypothesis of cutaneous plasmacytosis.
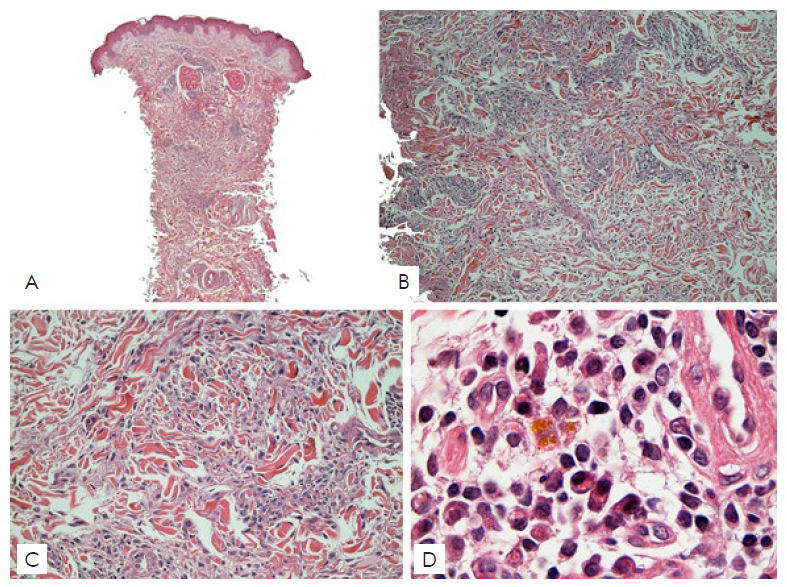
Figure 4 Assembly of 4 photomicrography images, in H&E stain, showing, in 4A, 4B and 4C, Interstitial Granulomatous Dermatitis pattern. 4D, in a 100x objective, detailing the massive presence of plasma cells in the infiltrate.
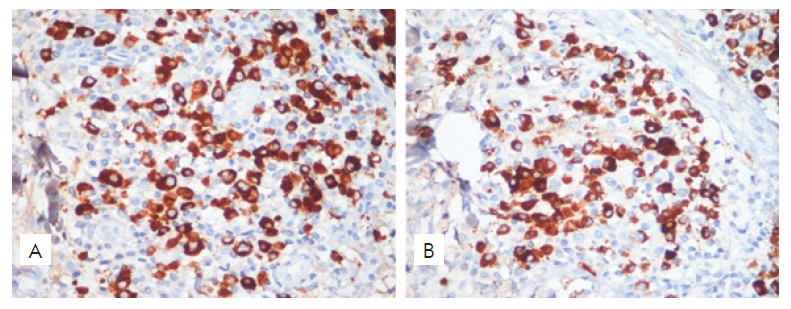
Figure 5 Polyclonality of infiltrating plasma cells of both κ- and λ-chain-positive cells was demonstrated with immunohistochemistry (400x).
Approximately 12 months after this presumptive diagnosis there was no apparent worsening and still no signs of systemic involvement, therefore a new treatment protocol using thalidomide with phototherapy was initiated, still with almost no improvement after 20 sessions of narrow-band UVB (NB-UVB).
DISCUSSION
In 1976, Yashiro et al first describe a case of a patient with cutaneous lesions where the biopsy showed a dense perivascular infiltrate of mature plasma cells, without a detectable underlying disease, suggesting that it could be “a kind of plasmacytosis”.4
In the 80’s, other reports with similar cutaneous and histopatho-logical findings were reported but sometimes associated with lymphadenopathy and polyclonal hypergammaglobulinemia.5-7Among them, Aso et al, in freased interleukin 6, a cytokine with a relevant role in the differentiation of B cells to plasma cells. Infection by human herpesvirus 8 may be also implicated, but was not assessed in this patient.8,9
Taking into account the possibility of systemic symptoms, some authors prefer the term “cutaneous and systemic plasmacytosis” to account for the extracutaneous involvement. However, as defined, there should be no significant underlying disease.10,11Particularly in this case, systemic involvement has been ruled out after a long follow-up and extensive and repeated clinical, laboratory and image exams. Therefore, we tend to consider it as a cutaneous isolated form, at least by now, but follow-up will be repeated at annual intervals.
Clinically, cutaneous plasmocytosis lesions are asymptomatic and progressive with multiple reddish-brown infiltrated macules, plaques and nodules, mostly on the trunk. The disease affects more often the elderly and Asians, unlike the case presented where the patient was a Caucasian with the lesional onset during childhood. In addition, the main site of reported involvement is the trunk, with progression to the upper limbs in those with disease for more than 2 years, which also differs from the case in which the lower limbs were the main areas affected. Axillary involvement is also reported in patients with long-standing skin disease, which was not observed in this patient.8
Clinical manifestations may include fever, anemia, lymphadeno-pathy and hypergammaglobulinaema.12 The patient in the present case had none of these manifestations.
Histopathology is characterized by the presence of moderately deep and superficial perivascular infiltrate of plasma cells without atypia. Immunohistochemistry shows polyclonal plasma cells with positivity for both κ and λ chains in similar proportions, with perineural distribution described in most cases.8,13Scleroderma is a relevant histopathological differential diagnosis, however clinically there was no sclerosis or atrophy. Infectious diseases, such as syphilis and borreliosis, could exhibit similar histopathological findings, however, serologies were non-reactive, in several occasions. Finally, the polyclonality of the infiltrate excludes a malignant neoplastic process. There is no standardized treatment for this disease, and most of the information was obtained from isolated case reports and small case series. Topical treatments include corticosteroids and calcineurin inhibitors. Systemic corticosteroids, thalidomide (probably associated with reduced IL6 secretion), ultraviolet-A (UVA) and narrow-band ultra-violet-B (NB-UVB) phototherapy (related to elevation of IL-10 with consequent IL6 suppression) are other options.8,14A good response was reported with 308-nm excimer lamp, with a mechanism supposedly similar to NB-UVB.15 The present patient has not responded to topical or systemic corticosteroid therapy neither to NB-UVB and thalidomide.
Despite the difficulty to achieve complete clearance with the available treatment options, the prognosis of the majority of patients with cutaneous/systemic plasmacytosis is favorable.8
CONCLUSION
With this illustrative case we aim to draw attention to this rare en-tity, discussing the relevant literature to the topic. We emphasize the importance of bringing clinical dermatology closer to pathology. Clinico-histopathological correlation was fundamental for the adequate diagnosis of this patient. This seems to be the first Brazilian report of Cutaneous Plasmacytosis recorded in the literature.

