











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
INTRODUCTION
Hailey-Hailey disease (HHD, OMIM 169600), also known as familial benign chronic pemphigus, was first described by the Hailey brothers in 1939.1 It is a rare autosomal dominant inherited disorder with an incidence of approximately 1 in 50 000 individuals.2 HDD first occurs after puberty, mostly in the third or fourth decade, with no sex or ethnic group predilection.2,3 Clinically, it presents as recurrent pruritic vesicles, painful erosions, and scaly erythematous plaques involving intertriginous areas and first occurring after puberty, mostly in the third or fourth decade.2,3Histopathologically, it is characterized by an intercellular split of the epidermal suprabasal layers (acantholysis) resulting from the disruption of cell-cell contacts.2,3Mutations in the ATP2C1 gene on band 3q22.1, encoding the secretory pathway Ca2+/Mn2+-ATPase protein 1(hSPCA1), have been identified as the cause of HHD.4 In human keratinocytes, hSPCA1 plays a significant role in maintaining calcium homeostasis between the cytoplasm and the Golgi apparatus, which is fundamental for maintaining desmosomal integrity.2Mutation in ATP2C1 might affect Ca2+ signaling in keratinocytes, altering the sorting of desmosomal proteins or their glycosylation, and resulting in the epidermal defects seen in skin lesions.2 Herein, we report the identification of two novel mutations of the ATP2C1 gene in two Portuguese patients.
CASE REPORTS
Case 1
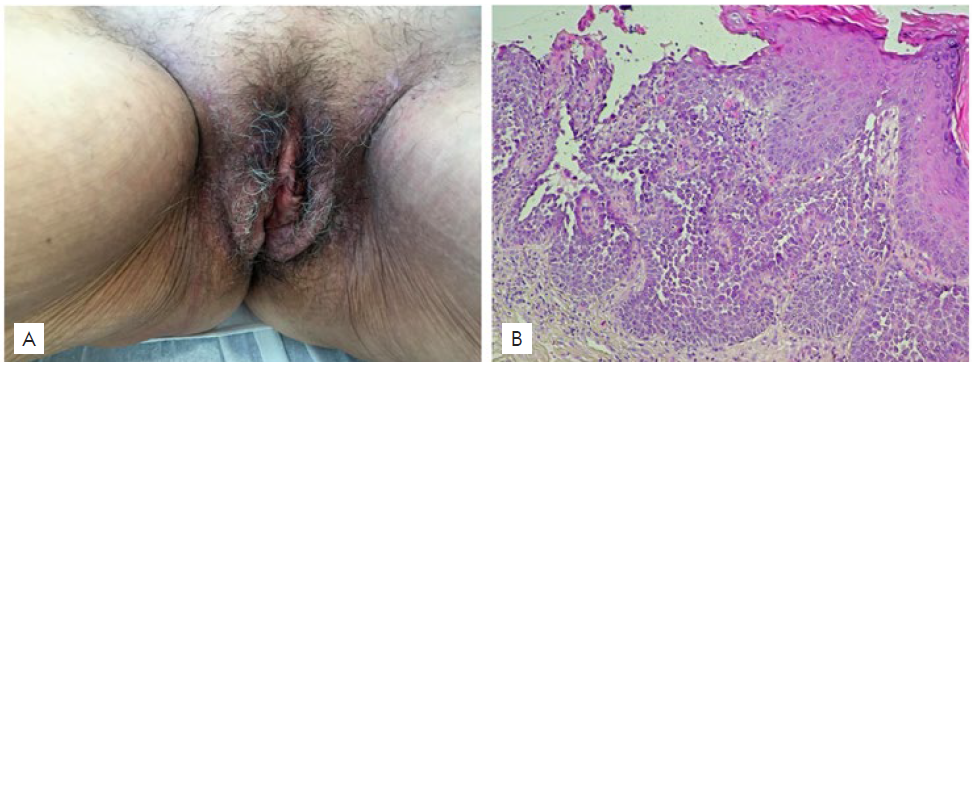
Patient 1 was a 56-year-old Portuguese Caucasian woman with a 2-year history of pruritic erythematous plaques on the groins and vulva, without previous diagnosis of HHD. Treatments with topical corticosteroid and fusidic acid, oral flucloxacillin and valaciclovir were used without any improvement of the skin lesions. Her mother had a history of similar skin lesions. Cutaneous examination revealed symmetrically distributed erythematous plaques with erosions and fis-sures on the groins and vulvar region (Fig. 1-A). Skin biopsy showed suprabasal keratinocyte acantholysis with mild dyskeratosis (Fig. 1-B). Accordingly, a diagnosis of HHD was established, and the pa-tient was successfully treated with oral acitretin 10 mg/day and topical corticosteroid, with total regression of the lesions within 5 months.
Case 2
Patient 2 was a 60-year-old Portuguese Caucasian woman with a 18-year history of typical skin lesions of HHD, mostly involving inframammary regions and groins (Fig. 2-A,B). The diagnosis of HHD was previously confirmed by histopathological study (Fig. 2-C,D). She reported 6 affected family members: the father, an uncle, a brother, a cousin, a niece and her son. The patient was admitted to the inpatient Dermatology Department for a clinical exacerbation of the skin lesions, already treated in the domicile with oral minocycline 100mg/day and betamethasone ointment, with minimal response. Bacterial culture from inguinal fold showed growth of Streptococcus pyogenes. The patient was treated with oral clarithromycin, oral acitretin 10 mg/day and zinc oxide oint-ment, with significant clinical improvement within 2 months.
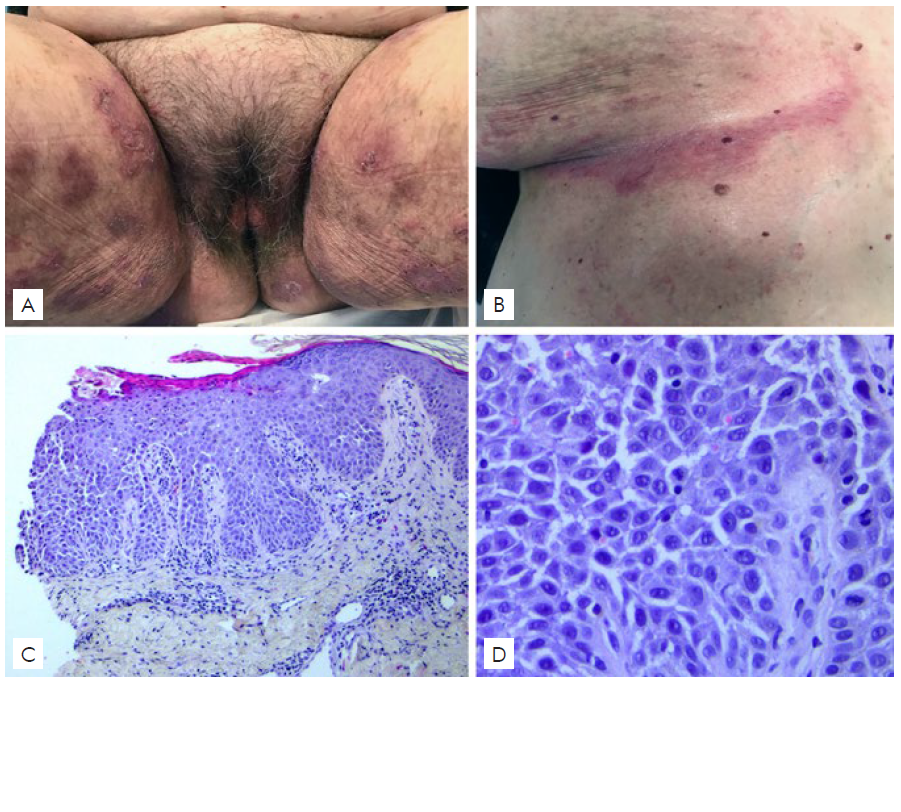
Figure 2 Patient 2(A,B) Clinical picture showing erythematous eroded involving the groins, perineum and inframammary folds. (C,D) Histological examination showing focal acantholysis beginning in the suprabasilar area and extending throughout the epidermis. (Hematoxylin and eosin, original magnification ×100, x400).
Genetic tests
After informed consent, genomic DNA was extracted from the patients’ peripheral blood using a standard in-house salting-out method.
A whole exome library was prepared using the Twist human core exome kit (Twist Bioscience). Target regions were sequenced (paired-end) on an Illumina platform (NovaSeq 6000) with 150 base read length, with a medium read depth of at least 60x. All the exonic and splice-site regions of the ATP2C1 gene were analyzed. To assess the potential functional consequences of detected variants and predict the clinical impact, bioinformatic data analy-sis was performed using well-established computational methods based on evolutionary conservation, protein structure and/or sequence homology.
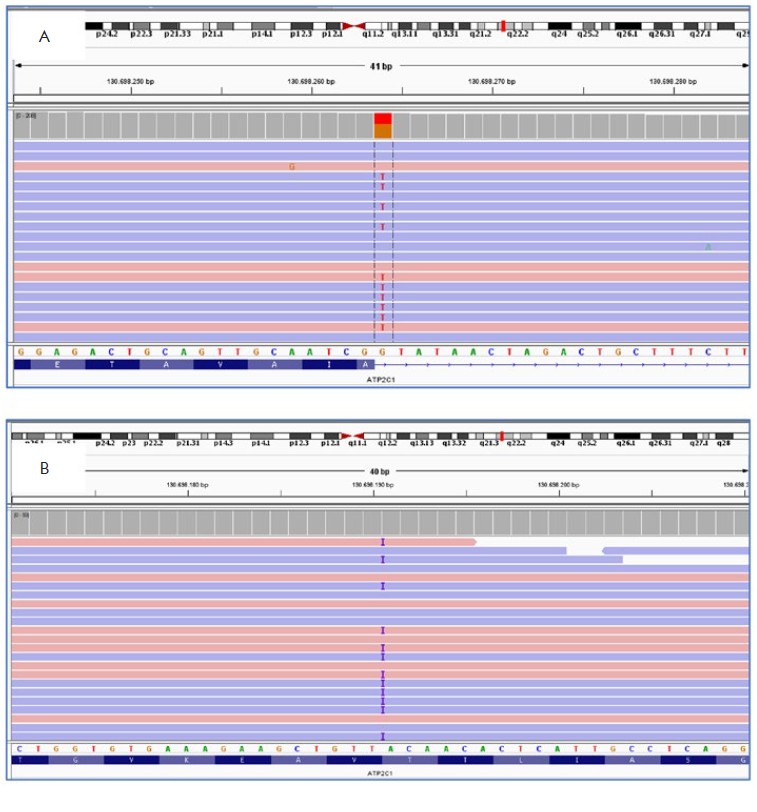
Two heterozygous mutations of the ATP2C1 gene were identified in these patients with HHD (Table 1). In patient 1, a donor splice site G>T transition at nucleotide 1741+1 (c.1741+1G>T) was found in intron 18 of ATP2C1 (Fig. 3-A). In patient 2, a duplication of an A at nucleotide 1669 was found in exon 18 of ATP2C1 (c.1669dupA), which altered the coding sense from Threonine to Asparagine at position 557, and creating a premature stop codon at position 65 [p.(Thr557Asnfs*65)] (Fig. 3-B). These two mutations were absent in public databases. They were classified according to the American College of Medical Genetics and Genomics guidelines as likely pathogenic.5
Table 1 Two novel mutations of ATP2C1 gene identified.
| Patient | Location | Nucleotide substitution | Aminoacid substitution | Mutation type |
| 1 | Intron 18 | c.1741+1 G>T | - | Donor splice site |
| 2 | Exon 18 | c.1669dupA | p.Thr557 Asnfs*65 | Frameshift |
DISCUSSION
Since ATP2C1 was first reported as the causative gene for HHD in 2000, at least 185 ATP2C1 gene mutations have been discovered worldwide.6 These mutations are scattered throughout the ATP2C1 gene, distributed all over the encoded sequence (27 exons) and in the intron splice sites, with no hotspots or clusters.7 They result in conformational defects and decreased levels of hSPCA1, leading to Golgi apparatus fragmentation, impaired cell proliferation, and defective sorting of proteins.6
No clear correlation between the genotype and phenotype was reported so far.7,8Age of onset, severity, or progression could not be attributed to the mutation location or type in the putative protein structure.8 Modifying genes and environmental factors may greatly influence clinical features of the disease.8
In summary, we identified two novel heterozygous mutations in the ATP2C1 gene in Portuguese patients with HHD. This result expands the repertoire of ATP2C1 mutations associated with HHD and provides useful information for genetic counseling for HHD patients and families. Further research is required to clarify the genotype-phenotype correlation.
Apresentações/Presentations
A paciente 1 foi apresentada em poster no 1º Congresso Virtual de Dermatologia e Venereologia, realizado de 21 a 22 de novembro de 2020, vencedor do prémio de melhor poster.

