











 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkDa citogenética à genómica: a evolução das tecnologias de investigação e diagnóstico
As tecnologias aplicadas à citogenética e à genómica têm evoluído muito ao longo dos anos, desde os primórdios da citogenética convencional até à moderna sequenciação massiva.
Citogenética convencional
A citogenética convencional ou análise do cariótipo diz respeito ao estudo do complemento cromossómico de um organismo, sendo os cromossomas corados de modo a evidenciar um padrão característico de bandas. A citogenética humana deu os primeiros passos em 1882, com a primeira ilustração de cromossomas humanos. Contudo, só em 1956, Tijo e Levan estabeleceram de forma correta e inequívoca que o complemento cromossómico humano normal é composto por 46 cromossomas (Tijo & Levan 1956). Três anos mais tarde foi identificada a primeira doença cromossómica, a trissomia 21, quando Lejeune, Gautier e Turpin observaram um pequeno cromossoma extra em metafases de fibroblastos provenientes de indivíduos com Síndrome de Down (Lejeune, Gautier & Turpin 1958). Surgiu no mesmo ano a identificação da Síndrome de Turner com o complemento 45,X (Ford, Jones, Polani, De Almeida & Briggs 1959) e a Síndrome de Klinefelter com 47,XXY (Jacobs & Strong 1959). Em 1960, Patau diagnosticou a trissomia 13 e Edwards a trissomia 18 (Edwards, Harnden, Cameron, Crosse & Wolff 1960; Patau, Smith, Therman, Inhorn & Wagner 1960). Foram assim reportadas as primeiras associações entre uma alteração cromossómica numérica e uma patologia. Ainda em 1960, no campo oncológico foi identificado o cromossoma Philadelphia em sangue periférico de doentes com leucemia mielóide crónica (Nowell & Hungerford 1960). Atualmente, várias alterações cromossómicas numéricas e estruturais têm sido descritas nos cromossomas sexuais e autossomas, verificando-se uma correlação direta com várias síndromes e neoplasias.
Num estudo citogenético, os cromossomas são identificados pelo seu tamanho, posição do centrómero e pelo seu característico padrão de bandas adquirido através de técnicas de coloração específicas. Os métodos de coloração ou bandagem dividem-se essencialmente em dois tipos: a) coloração diferencial de estruturas específicas do cromossoma e b) coloração de todo o cromossoma. São exemplo do primeiro tipo a bandagem-C, que visa as regiões de heterocromatina constitutiva presentes nos centrómeros, braços curtos de cromossomas acrocêntricos e regiões polimórficas dos cromossomas 1, 9, 16 e Y e, a bandagem NOR, que visa as regiões NOR (do inglês Nucleolar Organizing Region) presentes nos satélites dos braços curtos dos acrocêntricos (Figura 1A). A bandagem-G, também designada por GTG, é exemplo do segundo tipo de coloração e é o método de bandagem mais utilizado nos laboratórios de Citogenética (Figura 1B). Através deste método, os cromossomas apresentam uma coloração permanente e característica conferida pela ação da enzima proteolítica tripsina, seguida de coloração com o corante Giemsa.
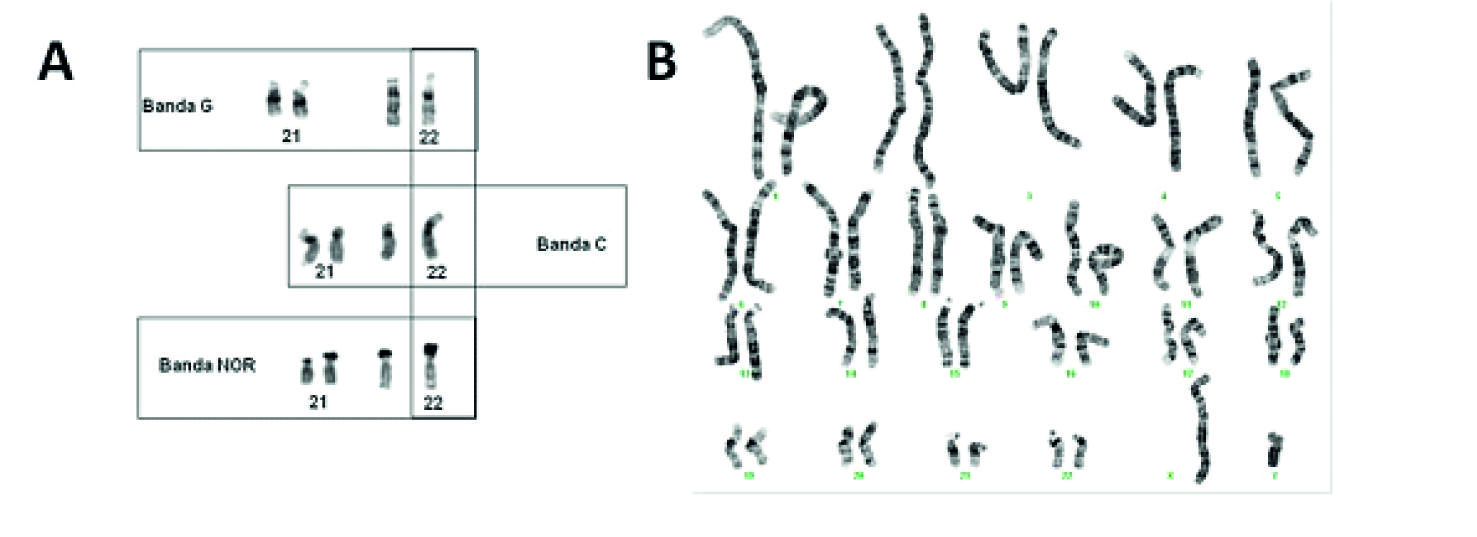
Figura 1 A) Exemplo de bandagem-G, C e NOR para os cromossomas 21 e 22. B) Cariograma humano com um padrão de bandas GTG de um indivíduo do sexo masculino com cariótipo 46,XY [ISCN 2016] (Imagens do Laboratório de Citogenética e Genómica da Faculdade de Medicina da Universidade de Coimbra - LCG-FMUC).
A obtenção de um padrão característico de bandas para cada cromossoma permite a sua identificação e o seu estudo detalhado, nomeadamente a deteção de alterações numéricas e estruturais. As alterações numéricas são erros no número de cromossomas considerado normal para uma determinada célula e as alterações estruturais são erros em que um ou mais cromossomas apresentam uma estrutura (padrão de bandas) anormal, como deleções, duplicações, translocações, inversões e inserções, entre outras. As alterações numéricas resultam de erros de segregação na divisão celular e são sempre desequilibradas, implicando perda ou ganho de um cromossoma. As alterações estruturais podem ser desequilibradas ou equilibradas, consoante está ou não envolvido ganho ou perda de material genético. Na maioria dos casos, as alterações estruturais equilibradas (translocações, inversões ou inserções), não estão associadas a manifestações clínicas para o portador; contudo, podem ser lesivas quando há disrupção de genes ou há alteração da expressão génica devido, por exemplo, a efeitos de posição. Adicionalmente, as alterações equilibradas podem ter graves consequências clínicas para a descendência dos portadores, resultando na formação de gâmetas com alterações cromossómicas desequilibradas. Estas alterações estruturais desequilibradas (deleções, duplicações, cromossomas dicêntricos, isocromossomas ou cromossomas em anel) estão frequentemente associadas a manifestações clínicas no portador.
As técnicas de citogenética convencional apresentam uma resolução relativamente limitada, permitindo a identificação de rearranjos estruturais superiores a cerca de 3 a 10 Mb (Keagle & Gersen 2005), não sendo detetadas alterações cromossómicas mais pequenas. O número de bandas observado depende do estado de condensação dos cromossomas, sendo que nos cromossomas metafásicos esse número atinge as 550 bandas enquanto que nos cromossomas prometafásicos, como apresentam menor condensação, o número de bandas é superior (ISCN 2009). Assim, a resolução da citogenética convencional depende de vários parâmetros como as características do microscópio, a qualidade das metafases e o modo como o DNA está compactado nos cromossomas (Li & Pinkel 2006).
Atualmente, a análise do cariótipo continua a ser uma ferramenta essencial para a genética clínica, uma vez que permite o estudo transversal de todo o genoma e de várias síndromes genéticas, facilmente diagnosticadas clinicamente e que estão relacionados diretamente com uma alteração detetada por citogenética convencional. Igualmente, os estudos citogenéticos permitem estabelecer o prognóstico e o acompanhamento de doentes oncológicos, uma vez que as alterações cromossómicas, nomeadamente as translocações, podem representar elementos-chave no desenlace clínico da doença oncológica. As alterações cromossómicas têm sido incluídas na classificação de leucemias, linfomas e tumores sólidos, complementando os critérios de prognóstico e diagnóstico (Wu, Sun, Zou & Chen 2007).
As principais vantagens da abordagem por citogenética convencional consistem no estudo simultâneo de todos os cromossomas, a deteção de alterações de ploidia e de alterações estruturais. As principais limitações são a resolução relativamente limitada e o facto de ser necessário a realização de culturas celulares, uma vez que a análise citogenética apenas se realiza em células em divisão celular mitótica.
Citogenética Molecular
A citogenética molecular permite o estudo de alterações genéticas utilizando tecnologias que combinam a citogenética convencional e técnicas moleculares (Speicher & Carter 2005). A introdução da citogenética molecular na prática clínica permitiu aumentar substancialmente o nível de resolução no diagnóstico dos rearranjos cromossómicos estruturais.
O princípio base das técnicas de citogenética molecular consiste no facto das sequências de nucleotídeos complementares hibridizarem entre si e formarem complexos mais estáveis do que as sequências não complementares (Speicher & Carter 2005). Em 1969 foi descrita a primeira experiência de hibridização in situ utilizando hibridização DNA-RNA para localizar os genes que codificam o RNA ribossómico, recorrendo a deteção radioativa (Gall & Pardue 1969).
Posteriormente, introduziu-se a marcação por fluorescência e as técnicas de citogenética molecular passaram a utilizar sondas de DNA marcadas com fluorocromos que hibridizam num alvo citológico que pode ser cromossomas em metafase, núcleos em interfase ou fibras de cromatina estendida (Speicher & Carter 2005).
Entre as técnicas de citogenética molecular temos a Fluorescence in situ hybridization (FISH) e a Comparative Genomic Hybridization (CGH). A técnica de Multiplex Ligation-dependent Probe Amplification (MLPA) é também muito utilizada em diagnóstico e permite a deteção de alterações no número de cópias de sequências cromossómicas específicas.
- Fluorescence in situ hybridization (FISH)
Na técnica de FISH a visualização do sinal resultante da hibridização de um fragmento de DNA marcado com um fluorocromo (sonda) à sua sequência complementar presente na amostra em estudo (DNA alvo) é realizada por microscopia de fluorescência. Esta deteção do sinal deve-se à propriedade de os fluorocromos serem excitados através de luz ultravioleta num comprimento de onda específico (l absorção) e, ao regressarem ao seu estado fundamental, emitirem luz num comprimento de onda maior e também específico (l emissão).
A metodologia de FISH tornou-se numa técnica fundamental para a deteção rápida e com elevada sensibilidade tanto de alterações numéricas como estruturais, em células quer em metafase quer em interfase. O tipo de sonda a usar depende do objetivo do estudo e da região alvo em análise, condicionando também a estratégia de hibridização a utilizar.
A sonda pode ser de marcação direta se um dos nucleotídeos incorporados na sonda já contém o fluorocromo, como por exemplo o 5-tiocianato de fluoresceína (FITC) que emite luz verde, a rodamina (TRITC) ou o Texas Red que emitem luz vermelha. Podem também ser de marcação indireta, se a sonda incluir um nucleotídeo associado a um hapteno, como a biotina ou a digoxigenina, que será detetado através de uma reação secundária com um anticorpo marcado com fluorescência (Volpi & Bridger 2008; Bishop 2010).
Após o Projecto Genoma Humano (International Human Genome Sequencing 2004) é, teoricamente, possível desenhar sondas de FISH para qualquer região cromossómica. As sondas de FISH produzidas comercialmente são sobretudo de três tipos:
1) Sondas de sequência única (ou locus-específicas) que podem ser geradas a partir de Bacterial Artificial Chromosome (BAC), P1 Artificial Chromosome (PAC), clones do tipo cosmídeo ou fosmídeo, ou por amplificação com primers específicos (Figura 2A), havendo também as sondas subteloméricas que são um grupo específico de sondas locus-específicas usadas para detetar rearranjos cromossómicos crípticos nas regiões terminais dos cromossomas (Figura 2C);
2) Sondas de sequências repetitivas que são constituídas por monómeros que se repetem em bloco várias centenas ou milhares de vezes, originando sinais fortes e facilmente visualizáveis, por exemplo específicas para as sequências satélite localizadas ao nível dos centrómeros (α-satélite) (Figura 2B) (de notar que os centrómeros humanos são distintos entre si, o que permite o desenvolvimento de sondas centroméricas específicas, excetuando os centrómeros dos cromossomas 13/21 e 14/22, que apresentam grande homologia); também se incluem nesta categoria as sondas para as sequências β-satélite (braços curtos dos cromossomas acrocêntricos), as sequências clássicas de DNA satélite (heterocromatina) e as sequências repetitivas teloméricas;
3) Sondas para pintura cromossómica que são geradas a partir de um conjunto de clones específicos para um cromossoma (ambos os braços, curto “p” e longo “q”, ou com marcação diferencial para cada um dos braços) e que permitem caracterizar e identificar cromossomas ou segmentos cromossómicos de origem desconhecida (Figura 2D) (Bishop 2010).
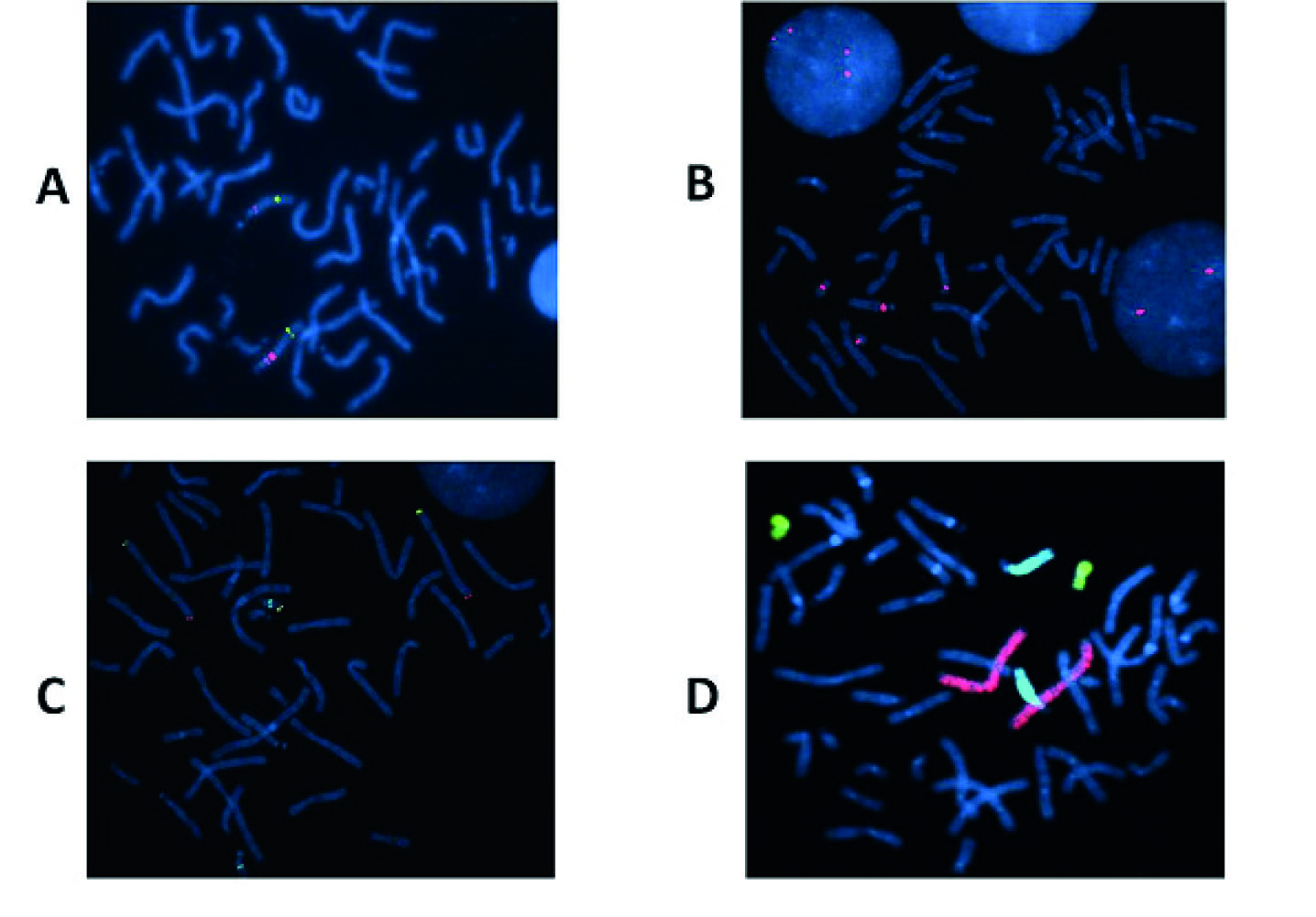
Figura 2 Técnica de FISH com diferentes tipos de sondas de DNA (marcação direta). Aplicação a metafases/interfases obtidas pela cultura sincronizada de linfócitos: (A) Sonda de sequência única: sonda SNRPN na região críptica 15q11.2, para a síndrome Prader-Willi/Angelman, evidenciando uma triplicação a vermelho. A verde está representada uma sonda controlo, mais distal, na região 15q26.3; (B) Sonda centromérica para os centrómeros dos cromossomas 13 (D13Z1) e 21 (D21Z1), a vermelho, em metafase e interfase; (C) Sondas para regiões subteloméricas dos cromossomas 3 e 22. Para o cromossoma 3: em 3p26.3, sonda D3S4559, a verde e, em 3q29, sonda D3S4560 a vermelho. Para o cromossoma 22: sonda MS607 em 22q13.33, a amarelo, combinada com sonda para 22q11.23 a azul claro, evidenciando uma deleção 22qter; (D) Sondas de pintura para os cromossomas: 2 (WCP2), a vermelho, 18 (WCP 18), a verde e 13 (WCP 13) a azul claro, evidenciando uma deleção no cromossoma 13 (Imagens do Laboratório de Citogenética e Genómica da FMUC).
A principal limitação da FISH é o facto de ser direcionada, obrigando à selecção de uma determinada região a estudar, estando limitada ao número de loci passível de ser testado em simultâneo ou a sucessivas hibridizações necessárias para completar o estudo. É, pois, uma técnica laboriosa e que não permite a avaliação de todo o genoma, sendo portanto utilizada principalmente para confirmar a presença de alterações cromossómicas já suspeitas. Contudo, apresenta uma resolução muito superior comparativamente à citogenética convencional (Riegel et al. 2005), é muito sensível, consegue detetar alterações cromossómicas em mosaico e pode ser realizada em vários tipos de tecidos, incluindo tecidos fixados em formalina e incluídos em parafina. No caso de se recorrer ao estudo de células apenas em interfase ou a amostras já fixadas, tem ainda a vantagem de permitir obter resultados em cerca de 24 horas. Alguns exemplos da aplicação de âmbito clínico desta tecnologia são a deteção de múltiplas cópias do gene HER2 em cancro da mama e do MYC em carcinomas e leucemias (Oliveira & French 2005), e na deteção rápida em diagnóstico pré-natal, em menos de 24 horas, das aneuploidias mais comuns (cromossomas 13, 18, 21, X e Y) em amniócitos não cultivados.
Desenvolveram-se algumas variantes da FISH, como o Multicolor FISH (M-FISH), o Spectral karyotyping (SKY) ou o fiber FISH, que permitem avaliações mais complexas dos cromossomas (Liehr et al. 2009). O M-FISH e o SKY permitem numa única experiência marcar cada par cromossómico com um espectro de cor diferente, utilizando várias combinações e proporções dos diferentes fluorocromos (Speicher & Carter 2005; Schrock et al. 1996). Estas variantes são muito úteis, pois permitem a caracterização de rearranjos complexos observados nas células tumorais, que envolvem translocações ou inserções e também, por exemplo, no estudo da origem de cromossomas marcadores, importante na etiologia do défice cognitivo. A grande desvantagem do SKY e do M-FISH é não detetarem inversões, deleções e duplicações intra-cromossómicas. Ainda, não permitem a determinação dos pontos de quebra exatos envolvidos nos rearranjos cromossómicos e é difícil determinar com certeza a origem do centrómero nos cromossomas derivativos de translocações (Liehr 2008). O fiber FISH utiliza fibras de cromatina estendidas, (l absorção) possibilitando a caracterização de rearranjos cromossómicos complexos (Heng, Squire & Tsui 1992; Wiegant et al. 1992) e a localização relativa e determinação da distância entre sequências adjacentes (Florijn et al. 1995; Giles et al. 1997).
- Comparative Genomic Hybridization (CGH)
A partir do princípio básico da técnica de FISH foi desenvolvida uma outra metodologia designada Comparative Genomic Hybridization (CGH) que permite o estudo abrangente e em simultâneo de todo o genoma. Nesta técnica, o DNA da amostra em estudo e de uma amostra de referência (diplóide), marcados com fluorocromos diferentes, são hibridizados conjuntamente em metafases de um controlo normal (diplóide) espalhadas numa lâmina de vidro. Esta co-hibridização competitiva permite, com recurso a um software adequado, calcular a razão de fluorescência para cada cromossoma e, assim, determinar o número de cópias de cada região cromossómica da amostra em estudo, comparativamente ao genoma de referência (Kallioniemi et al. 1992). Esta técnica foi desenvolvida inicialmente para detetar alterações no número de cópias em tumores sólidos. Tem como desvantagem a sua baixa resolução, idêntica à da citogenética convencional; contudo, tem a grande mais-valia de não necessitar do estabelecimento de culturas celulares, vantagem muito importante, nomeadamente nos estudos no âmbito da oncologia (Lichter, Joos, Bentz & Lampel 2000).
- Multiplex Ligation-dependent Probe Amplification (MLPA)
As alterações no número de cópias (ganhos/duplicações e perdas/deleções) de sequências cromossómicas específicas são frequentemente descritas como causa ou predisposição para doenças ou síndromes humanas (Schouten et al. 2002). Atualmente, diferentes técnicas podem ser utilizadas para a deteção de alterações no número de cópias de sequências cromossómicas, no entanto, muitas destas metodologias não são capazes de detetar perdas ou ganhos em exões únicos, e apresentam-se como técnicas morosas, difíceis de implementar como ensaios multiplex (como é o caso da FISH) ou requerem grandes quantidades de DNA (Schouten et al. 2002). Por exemplo, a Polymerase Chain Reaction (PCR) em tempo real fornece a possibilidade de detetar várias amplificações da sequência, mas a sua utilização em ensaios multiplex é bastante limitada pela sobreposição espectral dos corantes fluorescentes utilizados. Assim, a presença de múltiplos pares de primers em reações multiplex reduz a robustez da PCR e a consistência da quantificação (Schouten et al. 2002). Em 2002, Schouten e colaboradores (Schouten et al. 2002) mostraram que a técnica de Multiplex Ligation-dependent Probe Amplification (MLPA) é suficientemente sensível, reprodutível e específica para permitir a deteção de ganho ou perda de uma cópia de um único exão em amostras de DNA humano tão pequenas como 20 ng (3000 células/ 0,5 ml de líquido amniótico). A técnica de MLPA é um método semiquantitativo, que se baseia na hibridização e ligação de duas sondas adjacentes complementares à sequência alvo, de modo a detetar alterações no número de cópias ao nível genómico (ganhos ou perdas), em comparação com as amostras controlo. Nesta técnica são as sondas adicionadas às amostras que após hibridização com a sequência alvo serão amplificadas e quantificadas e não os ácidos núcleicos diretamente (Sellner & Taylor 2004) (Figura 3). Cada sonda de MLPA consiste em duas hemi-sondas de oligonucleotídeos, uma sintética e uma derivada do bacteriófago M13. Os dois oligonucleotídeos hibridizam em locais adjacentes da sequência-alvo, tendo cada hemi-sonda uma sequência de primer universal numa extremidade e uma das hemi-sondas uma sequência stuffer que permite a obtenção de produtos de PCR com tamanhos diferentes e únicos. Todas as sondas utilizadas num painel de sondas de MLPA são produzidas em diferentes vetores M13 e têm diferentes sequências stuffer e de hibridização (Schouten et al. 2002). Uma vez hibridizadas, as duas hemi-sondas são unidas por uma ligase termoestável e os produtos ligados são amplificados por PCR usando um único par de primers marcado. Os produtos de PCR de tamanho individual (130-480 pb) são depois separados por eletroforese capilar (Schouten et al. 2002). A quantidade relativa de cada produto de PCR é proporcional ao número de cópias da sequência-alvo, sendo os resultados apresentados como número de cópias do alelo em comparação com os controlos normais (Gouas, Goumy, Veronese, Tchirkov & Vago 2008).
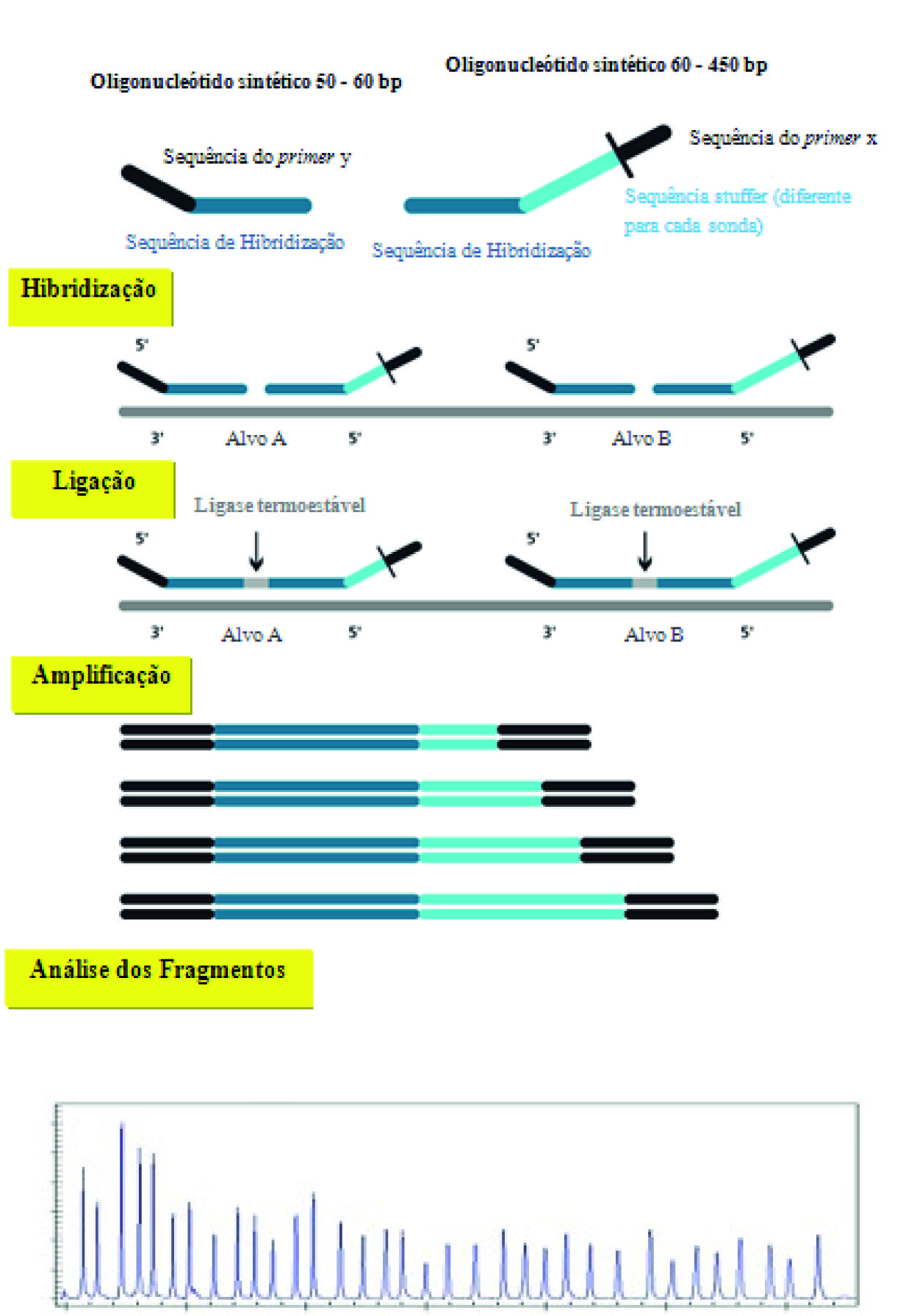
Figura 3 Esquema representativo das diferentes etapas da técnica de MLPA: 1) Hibridização das duas sondas adjacentes específicas para cada sequência alvo; 2) Reação de ligação pela enzima ligase; 3) Amplificação por PCR; 4) Separação por eletroforese capilar com respetiva análise dos fragmentos [adaptado de www.mgz-muenchen, acedido em novembro de 2010].
A técnica de MLPA tornou-se rapidamente muito popular, não só na investigação mas sobretudo como ferramenta de diagnóstico. As principais vantagens são o facto de utilizar uma pequena quantidade de DNA, não ser necessário cultura celular, permitir uma análise célere, poder ser utilizado DNA parcialmente degradado, como o DNA extraído de tecidos em parafina e fixados com formalina1, permitir o processamento de um número considerável de amostras em simultâneo e o estudo de um número considerável de loci para uma mesma região ou mesmo várias regiões/genes em simultâneo (dependendo do painel de sondas selecionado para o estudo). Adicionalmente, o equipamento necessário para a realização desta técnica (termociclador e sistema de eletroforese capilar) encontra-se presente na maioria dos laboratórios de biologia molecular, sendo por isso uma técnica que rapidamente se pode materializar (Schouten et al. 2002). Em oposição, as principais desvantagens são o facto de não detetar alteração de ploidias, mosaicismo de baixo grau e rearranjos equilibrados, apresentando também maior sensibilidade à contaminação e aos inibidores de PCR, como são exemplo os vestígios de fenóis. Adicionalmente, as mutações pontuais ou Single Nucleotide Polymorphism (SNP) que se encontram perto do local de ligação da sonda de MLPA podem afetar a eficiência da ligação da sonda, levando a uma aparente redução da altura relativa do pico e, consequentemente, a falsos positivos (Gouas, Goumy, Veronese, Tchirkov & Vago 2008).
Surgiram posteriormente várias variantes da técnica de MLPA com uma grande variedade de aplicações que incluem deteção de mutações (Scala et al. 2007) e de SNPs (Volikos et al. 2006), análise de metilação do DNA (Methylation-Specific MLPA (MS-MLPA) (Procter, Chou, Tang, Jama & Mao 2006) e quantificação relativa de mRNA (Wehner et al. 2005). Existem mais de 250 painéis de sondas comerciais para ensaios de MLPA com aplicações diferentes nas áreas do diagnóstico de doenças genéticas constitutivas ou adquiridas assim como na investigação2. Em termos de diagnóstico estes painéis são amplamente utilizados em laboratórios de genética clínica laboratorial.
Citogenómica
A citogenómica com a técnica de array-Comparative Genomic Hybridization (aCGH), foi inicialmente desenvolvida como uma ferramenta de investigação de desequilíbrios genómicos no âmbito da oncologia. A utilização de arrays de oligonucleotídeos para a análise do número de cópias com elevada resolução foi primeiramente descrita em 2003 por Lucito e colaboradores (Lucito et al. 2003) para detetar amplificações e deleções em amostras de cancro.
Atualmente, é uma ferramenta essencial e de rotina no diagnóstico genético, estando gradualmente a tomar o lugar dos métodos de citogenética convencional em vários laboratórios de genética38. Contudo, como as tecnologias citogenómicas não permitem detetar rearranjos equilibrados, é essencial a combinação da sua utilização com outras técnicas citogenéticas (convencional ou molecular), de modo a caracterizar convenientemente as amostras em estudo. O desenvolvimento e as aplicações clínicas das técnicas de citogenómica têm revolucionado o diagnóstico e facilitado a identificação das bases moleculares de várias doenças. É, hoje, o teste de primeira linha no estudo de doentes com défice cognitivo com ou sem dismorfismos e muito utilizada no diagnóstico pré-natal em fetos com malformações ecográficas. As metodologias usadas na citogenómica são o aCGH e o Single Nucleotide Polymorphism (SNP) array.
- Array-Comparative Genomic Hybridization (aCGH)
A técnica de aCGH, também designada por alguns autores como cariótipo molecular, é uma variante da CGH e que foi desenvolvida para a avaliação do genoma humano de modo a identificar desequilíbrios no número de cópias de DNA, detetando Variações do Número de Cópias (do inglês, Copy Number Variants - CNVs) (Solinas-Toldo et al. 1997). Com esta metodologia pode avaliar-se todo o genoma ou algumas regiões específicas. Tecnicamente, são co-hibridizadas num suporte sólido (na maioria das vezes uma lâmina de vidro) contendo várias sondas de DNA, iguais quantidades de DNA genómico do caso em estudo e da amostra controlo marcados com fluorocromos diferentes, nomeadamente Cyanine 5 (Cy5) e Cyanine 3 (Cy3). As sondas de DNA que se encontram no suporte físico podem ser fragmentos de DNA genómico humano clonados a partir de bactérias (BAC- Bacterial Artificial Chromosome de PAC - P1-derived Artificial Chromosomes), com um tamanho de cerca de 75-200 kb, ou ainda oligonucleotídeos sintéticos (25-85 mers), que actualmente são os mais utilizados (Shinawi & Cheung 2008). A resolução genómica das diferentes plataformas de aCGH é determinada pelo espaçamento e tamanho das sondas de DNA. Nas plataformas de aCGH para todo o genoma os alvos estão espaçados com uma cobertura de cerca de um clone por megabase a um clone por 100 Kb, mas podem ter cobertura variável consoante a região genómica (Veltman & de Vries 2006). Habitualmente, os arrays comerciais de oligonucleotídeos variam de uma sonda por 6 kb a uma sonda por 70 Kb. Assim, a resolução de um array cobrindo todo o genoma depende do número, distribuição e tamanho das sondas (Ylstra, Van Den Ijssel, Carvalho, Brakenhoff & Meijer 2006), dependendo da quantidade de regiões estudadas, do protocolo e da plataforma selecionados para o estudo. Na análise dos resultados, a intensidade da fluorescência é quantificada através de um scanner e com recurso a um software específico, sendo a razão resultante da intensidade de fluorescência entre a amostra em estudo e a amostra controlo, para cada locus, proporcional à razão do número de cópias do DNA em estudo comparativamente ao controlo. Deste modo, uma razão Cy5:Cy3 alterada é indicativa de perda ou ganho no DNA em estudo (Figura 4).
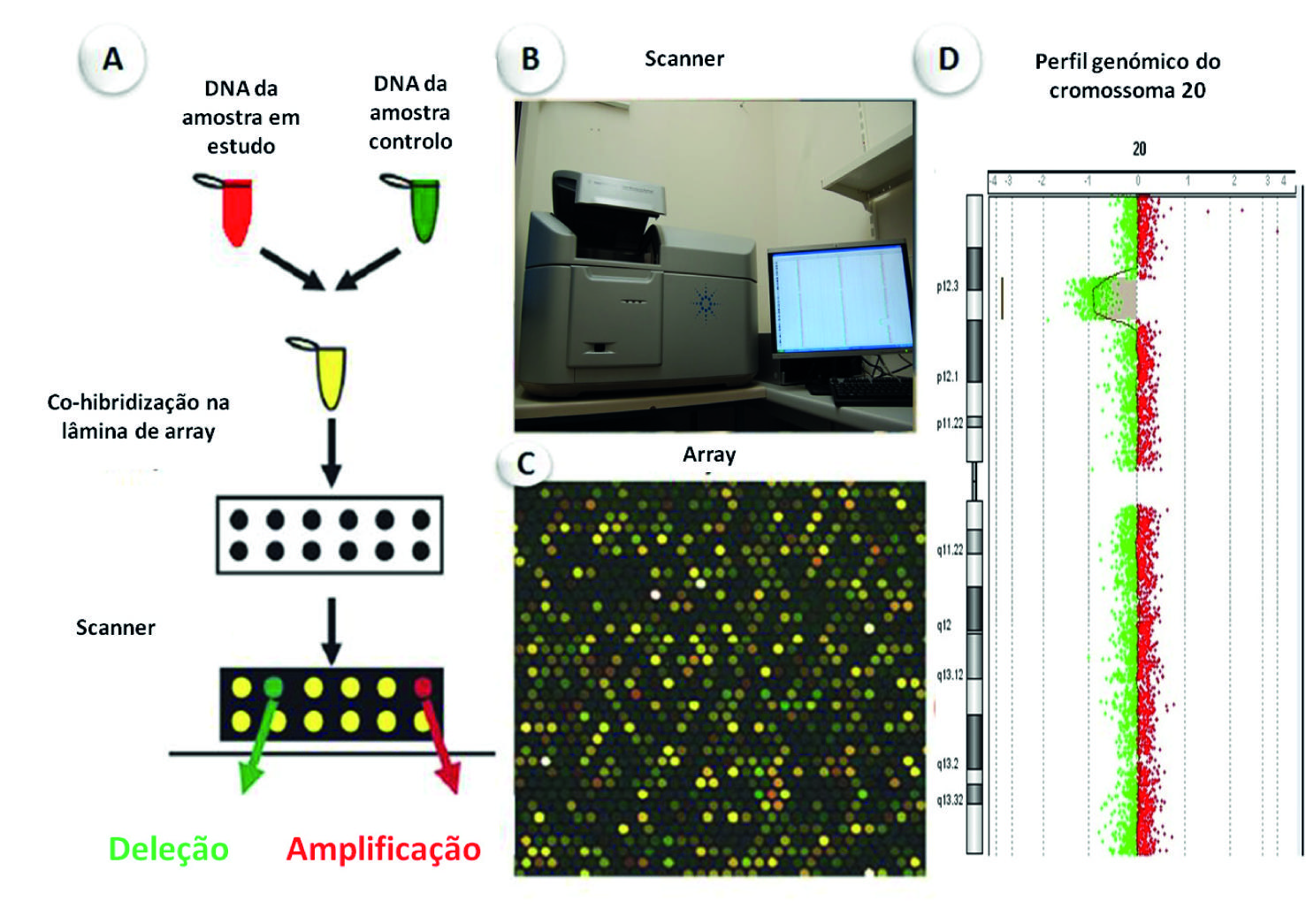
Figura 4 Esquema representativo da técnica de aCGH. A) O DNA da amostra a ser testada está marcado com fluorocromo vermelho (Cy5) e o DNA controlo está marcado com fluorocromo verde (Cy3). As duas amostras co-hibridizam competitivamente no array, que contém as sondas de DNA. As áreas que aparecem a vermelho na lâmina indicam material cromossómico extra (amplificação) na amostra em estudo na região em causa e, as áreas que aparecem a verde indicam menos DNA da amostra em estudo (deleção) na região em análise. B) As lâminas de array são lidas num scanner específico de microarray. C) Uma imagem obtida após leitura no scanner demonstrando diferentes razões de intensidade de fluorescência. D) Uma imagem exemplificativa do perfil genómico do cromossoma 20, obtida por aCGH. [adaptado de Veltman e Vries, 2006 (Shinawi & Cheung 2008)].
- Single Nucleotide Polymorphism (SNP) array
A plataforma de Single Nucleotide Polymorphism (SNP) array utiliza um tipo específico de oligonucleotídeos e foi inicialmente desenvolvida para estudos genómicos de linkage ou de associação em certas patologias. Esta tecnologia é semelhante à de aCGH, no entanto, neste caso não é feita hibridização comparativa e o suporte físico contém várias sondas para cada SNP do genoma que se pretende avaliar, o que permite a análise simultânea do número de cópias com a análise da perda de heterozigotia (LOH) ao nível do nucleotídeo, a realização de estudos de genotipagem e a avaliação de disomia uniparental e de consanguinidade (Speicher & Carter 2005; Bignell et al. 2004).
Genómica
Podemos dizer que o estudo genómico diz respeito ao estudo da sequência e análise do genoma de um organismo. As técnicas de sequenciação convencional, como por exemplo a técnica de Sanger, permitem apenas um estudo limitado e direcionado de uma determinada região do genoma. No entanto, existem atualmente metodologias que permitem a determinação da sequência de nucleotídeos de um modo mais abrangente.
A Next Generation Sequencing (NGS), ou sequenciação massiva em paralelo, designa a metodologia de sequenciação de milhões de pequenos fragmentos de DNA em simultâneo (Behjati & Tarpey 2013). Esta técnica permite a deteção de alterações da sequência de DNA relativamente à de referência, havendo várias plataformas disponíveis que utilizam diferentes tecnologias de sequenciação e que apresentam diferentes vantagens de acordo com o tipo de aplicação/objetivo do estudo (Metzker 2010; Liu et al. 2012; Kircher & Kelso 2010).
A NGS pode ser utilizada para sequenciar todo o genoma (whole genome sequencing), apenas as regiões codificantes dos genes (exome sequencing) ou um número limitado de genes (targeted sequencing). Atualmente, existe uma panóplia de variantes desta tecnologia, com diferentes aplicações, nomeadamente a técnica de RNAseq, que consiste na sequenciação de RNA para estudos de expressão genética (Wang, Gerstein & Snyder 2009; Wold & Myers 2008) e aplicações diferentes no campo da epigenética que permitem o estudo dos padrões de metilação e o estudo das interações entre proteínas e DNA e as modificações das histonas, utilizando a técnica de chromatin immunoprecipitation (ChIP) (Liu et al. 2012).
O rápido progresso das técnicas de sequenciação massiva tem permitido a redução dos seus custos e o aumento da sua precisão e, consequentemente, o alargamento das suas aplicações (Liu et al. 2012). De notar, também, que embora ainda com algumas limitações, existem já alguns algoritmos de análise associados à tecnologia de NGS que permitem a avaliação de CNVs ou a deteção de rearranjos cromossómicos equilibrados como inversões, inserções e translocações.
As metodologias por NGS estão em franco desenvolvimento e a sua aplicação nos laboratórios de genética clínica é cada vez maior e mais abrangente. Apresentam uma grande sensibilidade, permitindo a identificação de variantes mesmo em baixa percentagem de células (mosaicos de baixa expressão) (Behjati & Tarpey 2013). Existem vários trabalhos que mostram a sua aplicação inclusivamente na sequenciação do DNA de uma única célula (Gawad, Koh & Quake 2016). Antecipa-se, assim, a sua utilização, por exemplo, para identificar a presença de células tumorais em circulação em doentes, o que será crucial no contexto clinico de gestão do doente em oncologia. De notar que o uso destas tecnologias permite já a sequenciação de DNA fetal presente no sangue materno, aplicação que hoje em dia é já largamente utilizada em vários laboratórios (Dawson et al. 2013; Taglauer, Wilkins-Haug & Bianchi 2014).
No entanto, a NGS tem ainda algumas limitações, sendo a interpretação dos resultados ainda difícil em muitos casos, o que dificulta ou impossibilita, em algumas situações, a sua aplicação na clínica. O custo e a necessidade de equipamentos, quer de análise laboratorial quer de tratamento bioinformático, são ainda grandes entraves à utilização alargada da NGS nos laboratórios de genética clínica.
No âmbito da Oncologia
O cancro lidera as causas de morte a nível mundial, sendo um grave problema de saúde pública. É expectável que a sua incidência aumente devido ao crescimento e envelhecimento da população bem como ao estilo de vida associado a fatores de risco, como o álcool e o tabaco (Jemal et al. 2011). Deste modo, estima-se que em 2020 o número de novos casos por ano aumente para mais de 15 milhões a nível mundial, com o número de mortes anual associadas a atingir cerca de 12 milhões (Kanavos 2006). O cancro é frequentemente designado como uma doença do genoma, na medida em que resulta da aquisição de várias alterações ao nível do DNA, levando, por exemplo, à ativação de oncogenes (ganho de função) e inativação de genes supressores tumorais (perda de função).
O processo de carcinogénese é frequentemente descrito como um processo com múltiplas fases, onde as células normais são progressivamente e continuamente alteradas em células malignas devido à aquisição de propriedades específicas, nomeadamente as que lhes conferem capacidade de se dividirem sem controlo, de invadirem outros tecidos ou de escaparem à morte. O genoma das células tumorais pode conter várias alterações diferentes, como rearranjos cromossómicos, inserções ou deleções de segmentos de DNA, amplificação, alterações ao nível dos nucleotídeos e ainda a aquisição de novas sequências de DNA, provenientes de vírus (Stratton, Campbell & Futreal 2009).
Concomitantemente com as alterações genéticas, também são características das células neoplásicas as alterações epigenéticas - alterações na expressão dos genes sem modificação da sequência de DNA, nomeadamente a metilação do DNA, modificações de histonas e a regulação genética pós-transcripcional por RNAs não codificantes designados por microRNAs (Ducasse & Brown 2006). Em 1960, Peter Nowell e David Hungerford identificaram em doentes com leucemia mielóide crónica um pequeno cromossoma acrocêntrico com uma aparente deleção, o qual ficou conhecido por cromossoma Philadelphia (Nowell % Hungerford 1960). Com o advento das técnicas de bandagem, Janet Rowley verificou que o cromossoma Philadelphia não resultava de uma deleção mas sim de uma translocação recíproca entre o braço longo do cromossoma 9 e o cromossoma 22 (Rowley 1973). Posteriormente, com o recurso à citogenética molecular FISH constatou-se que da translocação entre os cromossomas 9 e 22 resulta um gene de fusão, o gene BCR-ABL (Heisterkamp et al. 1983; Groffen et al. 1984). Atualmente, existem mais de 63 mil rearranjos cromossómicos e mais de 1500 genes de fusão identificados em diferentes tipos de neoplasias humanas (Mitelman, Johansson & Mertens 2013).
Apesar dos progressos tecnológicos observados, nomeadamente com a introdução de tecnologias como o aCGH e a NGS, as técnicas de citogenética convencional e molecular continuam a ser fundamentais para o diagnóstico, o prognóstico e o tratamento de várias neoplasias, na medida em que permitem identificar rearranjos cromossómicos equilibrados e detetar mosaicos de baixa expressão. A amplificação do gene MYC em neuroblastomas, detetada por rotina pela técnica de FISH usando sondas de sequência única (locus-specific), está associada à agressividade da doença, sendo, portanto, uma técnica eficaz para auxiliar na estratificação do risco e na seleção do regime terapêutico nestes doentes (Dolan 2014).
Do mesmo modo, a amplificação do gene HER2 (ERBB2) é detetada em cerca de 10-30% dos casos de cancro da mama invasivo, sendo associada à agressividade da doença e também à resposta a agentes quimioterapêuticos específicos, sendo, portanto, a sua detecção fundamental para auxiliar na abordagem clínica destes doentes, que podem, por exemplo, beneficiar de terapias-alvo dirigidas para o gene HER2 (Dolan 2014).
É também importante referir que as perdas e os ganhos de material genético identificadas por outras técnicas, como o aCGH, podem ser validadas usando a técnica de FISH, que poderá depois ser usada para, por exemplo, monitorizar a evolução da doença no decurso dos tratamentos.
A vasta gama de painéis de sondas de MLPA para a caracterização molecular de amostras tumorais disseminou a sua utilização para a identificação de ganhos e perdas de material genético associados à progressão tumoral e à resposta terapêutica (Stuppia, Antonucci, Palka & Gatta 2012). A introdução da técnica de aCGH na investigação oncológica propiciou a identificação de várias regiões cromossómicas e novos genes associados a tumores específicos. Devido à elevada resolução desta técnica, foi possível verificar que translocações consideradas equilibradas pela citogenética convencional envolviam, afinal, ganho e perda de material genético (Watson, deLeeuw, Horsman, Squire & Lam 2007). Os avanços no conhecimento da genética do cancro introduzidos pela técnica de aCGH são bem patentes, uma vez que permitiram identificar padrões de alteração de ganho e perda de material genético, específicos para diferentes tipos de neoplasias (Jong et al. 2007; Bergamaschi et al. 2006) e associar essas alterações genéticas com as características clínico-patológicas dos doentes, nomeadamente a progressão tumoral, o prognóstico, a resposta terapêutica e a evolução clínica do doente (Kallioniemi 2008).
Com o advento das novas técnicas de sequenciação do genoma assiste-se a uma mudança no paradigma da investigação clínica em oncologia. Antecipa-se que a metodologia de NGS será importante para a monitorização do curso da doença oncológica através da deteção de alterações genéticas tumorais presentes em DNA no sangue periférico dos doentes (McBride et al. 2010). A avaliação da resposta à terapêutica de modo a detetar a aquisição de resistência é também uma aplicação que pode ser introduzida nas estratégias de tratamento dos doentes oncológicos (Meldrum, Doyle & Tothill 2011).
É indiscutível que a NGS permitiu, num curto espaço de tempo, avanços significativos no campo oncológico, nomeadamente através dos projetos como o The Cancer Genome Atlas (TCGA) (2008), o Tumor Sequencing Project (TSP) (Ding et al. 2008), e o The Cancer Genome Anatomy Project (Greenman et al. 2007), que visam sequenciar milhares de doentes com vários tipos de cancro, identificando, assim, novas mutações e genes sem associação conhecida com as neoplasias em estudo.
Apesar do progresso no conhecimento das bases moleculares envolvidas no processo de carcinogénese e do progresso tecnológico e avanços terapêuticos, o cancro continua a ser uma doença letal em todo o mundo, sendo responsável por inúmeras mortes (Garcia et al. 2007). A identificação de alterações genéticas e padrões epigenéticos associados ao desenvolvimento e progressão de cada neoplasia humana específica tem-se revelado importante na descoberta de novos biomarcadores de diagnóstico e de prognóstico. A validação destes biomarcadores permitirá a sua aplicação na prática clínica com benefícios na morbilidade e mortalidade associadas ao cancro (Ribeiro & Carreira 2014).
É, portanto, expectável que nos próximos anos através da aplicação e progresso de tecnologias genómicas haja um avanço significativo do conhecimento do processo de carcinogénese, com consequente translação à prática clínica (Corless 2016).
No âmbito das patologias do neurodesenvolvimento
As perturbações do neurodesenvolvimento constituem alterações neurobiológicas no desenvolvimento normal do feto e/ou da criança, podendo ser diagnosticadas ao nascimento ou manifestarem-se mais tarde, afetando a cognição, a comunicação, a linguagem, a motricidade, a atenção, a socialização, o comportamento, a aprendizagem escolar e a autonomia. Muitas das alterações celulares responsáveis por estas patologias ocorrem numa fase precoce do desenvolvimento, nomeadamente durante a fase pré-natal. Na categoria das perturbações gerais do desenvolvimento neurológico encontram-se o défice intelectual e as perturbações no espectro do autismo (Matos 2009; Ribeiro, Freitas & Oliva-Teles 2013).
Estes dois grupos de distúrbios do neurodesenvolvimento cruzam-se frequentemente nas suas manifestações clínicas e na sua etiologia, tendo muitas vezes origem em alterações genéticas e citogenéticas. De entre as causas genéticas mais frequentes encontramos as alterações cromossómicas, como por exemplo: as aneuploidias, responsáveis por síndromes, como a Síndrome de Down (trissomia 21) ou a Síndrome de Turner (monossomia do X) e as microdeleções e microduplicações associadas a síndromes de genes contíguos, como a Síndrome de DiGeorge (del(22)(q11.2)), a Síndrome de Miller Dicker (del(17)(p13.3)), a duplicação 15q11q13, a microdeleção 22q13, a microdeleção/duplicação 16p11.2, entre outras. Outro tipo de causa frequente são as alterações monogénicas nos autossomas e ligadas ao cromossoma X, como por exemplo aquela que ocorre na síndrome de X-frágil (Ferreira et al. 2013).
O diagnóstico precoce destas patologias, muitas vezes ainda em ambiente pré-natal, é fundamental para o adequado acompanhamento, apoiando e auxiliando os familiares, a equipa clínica e os educadores (Matos 2009; Ribeiro, Freitas & Oliva-Teles 2013). Com o recurso às várias técnicas de citogenética convencional, molecular e de genómica, têm-se verificado avanços significativos no diagnóstico tanto a nível de resolução tecnológica como nos tempos de resposta (é comum em 24 horas fazer-se o diagnóstico das aneuploidias mais comuns, 21, 13, 18 e dos cromossomas sexuais, tanto em pré como em pós-natal).
As diferentes abordagens tecnológicas têm permitido elucidar as bases biológicas destas perturbações do neurodesenvolvimento, que se refletem na identificação crescente de mutações (alterações da sequência normal das bases de DNA), e de CNVs (variações do número de cópia de um mesmo segmento de DNA, variando em tamanho, entre a ordem de 1 Kb a vários Mb, e podendo corresponder a regiões do genoma humano que incluem vários genes).
As alterações estruturais, como os rearranjos desequilibrados, nomeadamente cromossomas derivativos de translocações ou inserções, cromossomas recombinantes, deleções, duplicações, cromossomas marcadores, e as alterações numéricas podem ser detetadas pela citogenética convencional. Esta abordagem permite o diagnóstico em 10-15% dos indivíduos com perturbações do neurodesenvolvimento (Rauch et al. 2006; Ropers 2008). As técnicas de citogenética molecular, como a FISH e o MLPA permitem detetar de um modo rápido e preciso alterações submicroscópicas, aumentando assim a resolução e o número de desequilíbrios genómicos identificados em aproximadamente mais 5% dos casos (Rauch et al 2006), especialmente nas síndromes de microdeleção e microduplicação como as síndromes de Williams-Beuren, DiGeorge, Smith-Magenis e Miller-Diecker.
A deteção de rearranjos das regiões subteloméricas, nomeadamente por FISH, também aumentou o diagnóstico em 2.5-5 % dos casos com défice intelectual (Knight et al. 1999). Mais recentemente, a tecnologia de aCGH permitiu a descoberta de um grande número de novas síndromes de microdeleção e microduplicação associados a estes distúrbios do desenvolvimento (Shaffer et al. 2007; Slavotinek 2008). Atualmente, em termos de estratégia de diagnóstico clínico, a técnica de aCGH tem sido considerada como o teste genético de primeira linha para detetar alterações genómicas em doentes com défice intelectual não sindrómico, com ou sem dismorfismos, anomalias congénitas múltiplas, dificuldades de aprendizagem e/ou distúrbios no espectro autista (Carreira et al. 2015; Miller et al. 2010).
Esta técnica apresenta maior resolução, permitindo assim a deteção de desequilíbrios não detetados pela citogenética convencional e, portanto, o número de diagnósticos destas patologias do neurodesenvolvimento sofreram um aumento em cerca de 10% (Nicholl et al. 2014). A utilização da NGS no estudo etiológico destas patologias constitui ainda um desafio, devido à necessidade de poderosas ferramentas bioinformáticas para analisar e interpretar o grande número de novas variantes que são identificadas. Nos últimos anos e com recurso à NGS, a identificação da etiologia destas perturbações do neurodesenvolvimento tem sofrido avanços significativos, tendo ajudado a identificar centenas de genes associados ao neurodesenvolvimento (Carvill & Mefford 2015).
Contudo, a etiologia da maioria dos casos de défice cognitivo e de perturbações no espectro do autismo permanece ainda por caracterizar, antecipando-se que muitos genes relacionados com estas patologias ainda sejam identificados no futuro (Carvill & Mefford 2015). A título de exemplo, e num estudo recente realizado pelo consórcio Deciphering Developmental Disorders (2015), verificou-se que a aplicação combinada de exome sequencing e aCGH em 1133 crianças com perturbações severas do desenvolvimento sem diagnóstico molecular permitiu a identificação de 12 novos genes associados a estas perturbações, o que se traduziu nesta amostra num aumento em cerca de 10% da capacidade diagnóstica. Estes resultados mostram a importância de estratégias combinadas utilizando diferentes tecnologias de citogenética e genómica para melhorar o conhecimento atual da etiologia das perturbações do neurodesenvolvimento e, consequentemente, do seu diagnóstico. A identificação destas alterações genéticas permite contribuir para a compreensão do mecanismo biológico associado e, consequentemente, para calcular o risco de recorrência numa próxima gravidez, bem como orientar familiares de risco para consultas de genética médica e estudos de genética clínica laboratorial. De notar, ainda, que a existência de equipas multidisciplinares é de fulcral importância, sobretudo na gestão e na decisão em casos complexos.
As diferentes tecnologias de citogenética e genómica atualmente disponíveis podem ser usadas em conjunto, complementando-se e ultrapassando as suas limitações individuais. Existem várias técnicas que permitem uma caracterização genética abrangente de todo o genoma e, por outro lado, outras técnicas que permitem o estudo direcionado de algumas regiões cromossómicas ou genes. A seleção das técnicas e protocolos a usar depende do objetivo do estudo, sendo sempre necessário a correlação genótipo-fenótipo para uma correta análise dos resultados, quer em estudos no âmbito do neurodesenvolvimento quer na área oncológica. No futuro, recorrendo também à sequenciação massiva em simultâneo e com a melhoria da capacidade de interpretação dos resultados, antecipa-se um papel cada vez mais importante do estudo citogenético e genómico em diferentes áreas clínicas.

