










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista Portuguesa de Medicina Geral e Familiar
versão impressa ISSN 2182-5173
Rev Port Med Geral Fam vol.30 no.6 Lisboa dez. 2014
ARTIGOS BREVES
Criança com hipertrofia gemelar e elevação das transaminases: relato de caso
Calf hypertrophy and raised transaminases in a child: a case report of muscular dystrophy
Joana Extreia,1 Paula Afonso,1 Margarida Rafael,1 Susana Rocha2
1 Médica interna de Pediatria.
2 Assistente Hospitalar de Pediatria
1, 2 Serviço de Pediatria, Centro Hospitalar Barreiro-Montijo
Endereço para correspondência | Dirección para correspondencia | Correspondence
RESUMO
Introdução: As transaminases não são enzimas específicas do hepatócito e são várias as condições que se associam ao seu aumento. A Distrofia Muscular de Duchenne (DMD) é uma doença de transmissão autossómica recessiva ligada ao cromossoma X que se caracteriza por fraqueza muscular progressiva.
Descrição do caso: Apresentamos o caso de um menino de 6 anos referenciado à consulta de pediatria para investigação de elevação persistente do valor das transaminases. Sem história familiar de doença hepática ou neuromuscular. Dos antecedentes pessoais a salientar, marcha autónoma aos 15 meses, tendência para marcha em pontas, dificuldade em subir escadas e correr e cansaço fácil em relação aos pares.
À observação destacava-se hipertrofia gemelar e abdominal, ligeira hiperlordose lombar, diminuição da força muscular proximal dos membros inferiores e manobra de Gowers positiva. Perante a história clínica, exame objetivo e exames complementares já realizados, foi colocada a hipótese de distrofia muscular e doseada também a enzima creatina quinase (CK) cujo valor estava muito aumentado. A biópsia muscular evidenciou a ausência de distrofina e a sequenciação do gene desta proteina identificou a mutação c.3103C>T (p.Gln1035X) confirmando o diagnóstico de DMD.
Comentário: Nem sempre o aumento das transaminases é sinónimo de doença hepática. Perante a elevação mantida destas enzimas, a exclusão de hepatopatia é obrigatória. No entanto, este achado laboratorial também pode surgir na doença muscular. Destacamos o doseamento da CK como avaliação mandatória, evitando a investigação exaustiva centrada na patologia hepática.
Os autores pretendem alertar para o diagnóstico diferencial de doença neuromuscular através de um caso em que a anamnese, a clínica e o perfil analítico são sugestivos.
Palavras-chave: Transaminases; Creatina Quinase; Distrofia Muscular; Hipertrofia Gemelar.
ABSTRACT
Introduction: Transaminases are non-specific markers of hepatocyte function. There are several conditions associated with elevated transa-minases such as Duchenne muscular dystrophy (DMD), an X-linked recessive disorder characterized by progressive muscle weakness.
Case Report: We report the case of a 6 year-old boy referred to the pediatric department for evaluation of raised transaminases. He had no family history of liver or neuromuscular disease. From his past history we noted independent gait at 15 months, toe-walking, difficulty climbing stairs and running, and fatigue in comparison to his peers. On clinical examination there was calf and abdominal hypertrophy, slight lumbar lordosis, proximal lower limb weakness and a positive Gowers’ sign. Given the clinical history, physical examination and la-boratory findings, the diagnosis of muscular dystrophy was suspected. Serum creatine kinase was found to be elevated. A muscle biopsy was performed and revealed absence of dystrophin. DMD gene sequencing identified the mutation c.3103C > T (p.Gln1035X) which confirmed the diagnosis of DMD.
Comment: The finding of raised transaminases is not always diagnostic of liver disease. With prolonged elevation of those enzymes, liver disease must be excluded but this laboratory finding may also appear in muscle disease. CK levels should also be measured to avoid exhaustive testing for liver disease. The authors advise clinicians to consider neuromuscular diseases in the differential diagnosis of cases presenting a with suggestive clinical and laboratory findings.
Keywords: Transaminases; Creatine Kinase; Muscular Dystrophy; Calf Hypertrophy.
Introdução
A alanina aminotransferase (ALT) e aspartato aminotransferase (AST) são enzimas intracelulares com papel fundamental no metabolismo dos aminoácidos. Embora seja no hepatócito que atingem maior concentração, estas enzimas encontram-se também noutros órgãos como o músculo cardíaco e esquelético. A ALT localiza-se exclusivamente no citoplasma e a AST encontra-se no citoplasma e na mitocôndria.1-2
Nas situações que cursam com lesão dos miócitos há libertação das respetivas enzimas intracelulares com elevação sérica das mesmas, incluindo as transaminases. A enzima creatina quinase (CK) vai estar concomitantemente elevada nestas situações, tornando a patologia hepática menos provável. As doenças neuromusculares cursam então com aumento da CK e das transaminases, entre outras enzimas da célula muscular como a aldolase, sendo que as alterações laboratoriais podem ser o primeiro indício para o diagnóstico em indivíduos assintomáticos.1-3
As distrofias musculares constituem um grupo de doenças musculares primárias, hereditárias, caracterizadas por fraqueza muscular progressiva, com grande variabilidade, quer fenotípica quer da sua gravidade. A transmissão é autossómica recessiva ligada ao X e distinguem-se vários tipos de acordo com a clínica. A Distrofia Muscular de Duchenne (DMD) é a forma mais grave e mais comum, com uma incidência de um em cada 3.500 recém-nascidos do sexo masculino.4-5
A DMD é provocada por mutações no gene da distrofina, uma proteína fundamental na manutenção da integridade da membrana da célula muscular. As mutações mais frequentes são as deleções e cerca de um terço ocorrem de novo.6 As manifestações clínicas incluem fraqueza muscular progressiva de atingimento inicialmente proximal e dos membros inferiores; dificuldade em correr, saltar e subir escadas são as queixas mais precoces com início entre os três e cinco anos de idade na maioria dos casos, associadas a pseudohipertrofia gemelar (secundária à infiltração do músculo por gordura e tecido conjuntivo). A lordose lombar e marcha miopática (tipo “pato”) são também características do quadro clínico. Alguns doentes caminham em pontas por contraturas ou retrações aquilianas. A manobra de Gowers é característica das patologias que cursam com fraqueza muscular proximal e é traduzida pelo apoio das mãos para elevação da posição sentada para a posição ortostática. Com a progressão da doença há envolvimento dos grupos musculares distais e também dos membros superiores. A fraqueza muscular progressiva associada ao agravamento da cifo-escoliose levam à degradação gradual da função respiratória. O músculo cardíaco vai estar envolvido em estadios mais tardios da doença e condiciona importante comorbilidade, como cardiomiopatia dilatada e alterações da condução cardíaca.4,6 Apesar de não existir ainda cura, são várias as intervenções terapêuticas que podem modificar a história natural da doença, nomeadamente a corticoterapia combinada com a fisioterapia, pelo que hoje em dia a maioria dos doentes atinge a idade adulta.7-9
Os autores pretendem alertar para o diagnóstico diferencial de doença neuromuscular, destacando a importância da clínica como principal pista para o diagnóstico.
Descrição do caso
Menino de seis anos e 10 meses que foi referenciado à consulta de pediatria geral pelo seu médico assistente por aumento persistente das transaminases.
Trata-se do primeiro filho de casal jovem, não consanguíneo, sem história familiar relevante, nomeadamente sem antecedentes de doença hepática ou neuromuscular. Gestação de termo, vigiada, parto eutócico, Índice de Apgar 4/6/10 ao 1º, 5º e 10º minuto, respetivamente. Somatometria ao nascer adequada e período neonatal sem intercorrências. Boa evolução estaturo-ponderal e do perímetro cefálico. Desenvolvimento psicomotor normal, com início da marcha autónoma aos 15 meses, embora com tendência a marcha em pontas e dificuldade em correr e subir escadas em comparação com os pares, com quedas frequentes. Referência, ainda pelos pais, a aumento de volume da região gemelar, notado desde cedo mas nunca valorizado pelos profissionais de saúde.
Na consulta de vigilância infantil aos cinco anos, por suspeita clínica de hepatomegália, na ausência de outros sinais sugestivos de hepatopatia, foi pedida a primeira avaliação laboratorial e imagiológica onde se destacava apenas o aumento das transaminases, com doseamentos de bilirrubina, fosfatase alcalina, gama-glutamiltransferase, albumina e tempo de protrombina normais. A ecografia hepática não tinha alterações, não se confirmando a suspeita clínica.
Perante esta alteração foram repetidas as análises e pedidas serologias víricas, que evidenciaram elevação persistente da ALT e AST sem alterações noutras provas de função hepática e com serologias víricas para os vírus da hepatite A, B, C, Epstein-Barr e citomegalovírus negativas. Pelo aumento mantido das transaminases, sem clínica sugestiva de hepatopatia, o doente foi encaminhado à consulta de pediatria hospitalar.
À observação destacava-se hipertrofia, simétrica e bilateralmente, dos músculos gemelares (figura 1) e abdominais, ligeira hiperlordose lombar, com diminuição da força muscular proximal dos membros inferiores e manobra de Gowers positiva. Retração aquiliana bilateral, impossibilitando a marcha em calcanhares. Anictérico, sem hepato ou esplenomegália, sem sinais de discrasia hemorrágica, sem distensão abdominal.
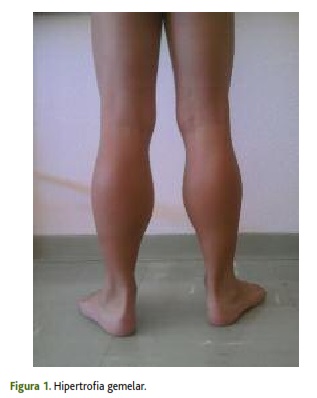
Com base na história clínica e alterações no exame objetivo foi colocada a hipótese de distrofia muscular e foram pedidos exames complementares de diagnóstico em concordância: o doseamento da CK mostrou elevação cem vezes superior ao valor normal, concomitante com aumento das transaminases, AST e ALT, ambas aumentadas dez vezes acima do valor de referência; a biópsia muscular evidenciou a ausência de distrofina. O estudo genético inicial foi no entanto normal, sendo apenas através da sequenciação do gene da distrofina que se identificou a mutação c.3103C>T (p.Gln1035X) em hemizigotia, confirmando o diagnóstico de DMD.
O doente foi encaminhado para um centro de referência de doenças neuromusculares onde mantém seguimento e os progenitores referenciados para aconselhamento genético. Atualmente, o doente está sob terapêutica com corticóide oral, realiza sessões de fisioterapia regulares, bissemanais, preservando ainda a capacidade de deambulação mas com agravamento progressivo da lordose lombar, não havendo ainda evidência de envolvimento cardíaco, comprometimento ventilatório ou articular.
Comentário
A alteração das transaminases pode surgir em várias situações para além da hepatopatia. A necessidade de investigação mais profunda e a urgência dessa mesma investigação constituem um desafio da prática clínica em qualquer idade.2
Para além do fígado, a ALT encontra-se nos músculos cardíaco e esquelético e a AST em órgãos como pulmão, rim e cérebro, embora em menor concentração.2,14 Já a CK também está presente no hepatócito, mas é no miócito que atinge maior concentração. É fundamental ter estas premissas em conta perante um doente com aumento mantido dos valores destas enzimas.14
A elevação isolada destas enzimas é muitas vezes um achado acidental em doentes assintomáticos e pode ser a primeira manifestação de uma hepatopatia grave ou de uma patologia extra-hepática. É fundamental que todos os clínicos tenham um elevado índice de suspeição, não só para miopatias, mas também para situações menos comuns que cursam com aumento das transaminases como cardiopatia, nefropatia, doença hemolítica e até doença celíaca;2 a história clínica deve ser orientada para a pesquisa de sintomas e sinais que os coloquem na direção diagnóstica correta, sendo fundamental valorizar devidamente a preocupação dos pais ou acompanhantes, não dando início intempestivo a uma investigação direcionada apenas à doença hepática.
Não considerar a hipótese de lesão muscular perante o aumento persistente das transaminases origina muitas vezes o atraso no diagnóstico de miopatias e perpetua a investigação direcionada para a patologia hepática, com realização de exames invasivos e dispendiosos.14
Vários autores descrevem séries de casos de atraso no diagnóstico de doença neuromuscular perante um aumento isolado e persistente das transaminases. Muitos doentes são inadequadamente referenciados a Unidades de Gastrenterologia e muitos deles submetidos a exames invasivos (nomeadamente biópsia hepática em alguns casos) com o objetivo de identificar patologia hepática inexistente. Há relatos de caso em que o atraso no diagnóstico foi superior a um ano.10-16 Na ausência de uma causa hepática identificada, testes simples como o doseamento da CK e aldolase devem ser realizados numa fase inicial para redirecionar a marcha diagnóstica.
Ao apresentar este caso clínico os autores pretendem destacar a importância de colocar causas extra-hepáticas, nomeadamente doença neuromuscular, como hipótese diagnóstica perante um aumento mantido das transaminases e também realçar a clínica que, neste caso, foi determinante para o diagnóstico. A idade, o sexo, os detalhes da anamnese, como dificuldade em subir escadas, marcha em pontas, quedas frequentes e o exame objetivo com manobra de Gowers positiva e hipertrofia gemelar fizeram colocar a hipótese diagnóstica de distrofinopatia e a partir daí orientar a investigação complementar para essa entidade nosológica, não persistindo na busca exaustiva de patologia hepática. O fator de confusão, neste caso, e o que terá atrasado a suspeita diagnóstica foi a palpação abdominal sugestiva de hepatomegália quando na verdade se tratava de uma pseudo-hipertrofia dos músculos abdominais.
A elevação das transaminases como achado acidental, até mesmo na ausência de clínica, pode ser uma oportunidade única e precoce de diagnosticar uma doença potencialmente grave ainda numa fase assintomática, permitindo assim a otimização terapêutica e a implementação de estratégias de reabilitação motora que melhorem a qualidade de vida dos doentes. Não menos importante é o aconselhamento genético atempado aos progenitores.
O prognóstico desta distrofinopatia é reservado, com perda da capacidade de deambulação no início da segunda década de vida, variando com a altura de início da corticoterapia. Esta demonstrou ser eficaz no prolongamento da capacidade para a marcha e atrasa a progressão da disfunção respiratória, cardiomiopatia e escoliose. A esperança média de vida situa-se, no entanto, na terceira década de vida. Atualmente estão a decorrer ensaios clínicos com terapia genética no sentido de se poder modificar a progressão natural da doença.
REFERÊNCIAS BIBLIOGRÁFICAS
1. Kohli R, Harris DC, Whitington PF. Relative elevations of serum alanine and aspartate aminotransferase in muscular dystrophy. J Pediatr Gastroenterol Nutr. 2005;41(1):121-4. [ Links ]
2. Lesmes Moltó L, Albañil Ballesteros MR. Aumento aislado de transaminasas: aproximación diagnóstica. Form Act Pediatr Aten Prim. 2013;6(1):35-42. [ Links ] Spanish
3. Vajro P, Maddaluno S, Veropalumbo C. Persistent hypertransaminasemia in asymptomatic children: a stepwise approach. World J Gastroenterol. 2013;19(18):2740-51. [ Links ]
4. Yiu EM, Kornberg AJ. Duchenne muscular dystrophy. Neurol India. 2008;56(3):236-47. [ Links ]
5. Monckton G, Hoskin V, Warren S. Prevalence and incidence of muscular dystrophy in Alberta, Canada. Clin Genet. 1982;21(1):19-24. [ Links ]
6. Reed UC. Doenças neuromusculares (Neuromuscular disorders). J Pediatr. 2002;78 Suppl 1:S89-S103. [ Links ] Portuguese
7. López-Hernández LB, Vázquez-Cárdenas NA, Luna-Padrón E. Distrofia muscular de Duchenne: actualidad y perspectivas de tratamento (Duchenne muscular dystrophy: current aspects and perspectives on treatment). Rev Neurol. 2009;49(7):369-75. Spanish
8. Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010;9(1):77-93. [ Links ]
9. Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: implementation of multidisciplinary care. Lancet Neurol. 2010;9(2):177-89. [ Links ]
10. Rutledge J, Andersen J, Fink CW, Cook J, Strickland A. Persistent hypertransaminasemia as the presenting finding of childhood muscle disease. Clin Pediatr. 1985;24(9):500-3.
11. Zamora S, Adams C, Butzner JD, Machida H, Scott RB. Elevated aminotransferase activity as an indication of muscular dystrophy: case reports and review of the literature. Can J Gastroenterol. 1996;10(6):389-93. [ Links ]
12. Korones DN, Brown MR, Palis J. "Liver function tests" are not always tests of liver function. Am J Hematol. 2001;66(1):46-8. [ Links ]
13. Urganci N, Arapoglu M, Serdaroglu P, Nuhoglu A. Incidental raised transaminases: a clue to muscle disease. Ann Trop Paediatr. 2006;26(4):345-8. [ Links ]
14. McMillan HJ, Gregas M, Darras BT, Kang PB. Serum transaminase levels in boys with Duchenne and Becker muscular dystrophy. Pediatrics. 2011;127(1):e132-6. [ Links ]
15. Wright MA, Yang ML, Parsons JA, Westfall JM, Yee AS. Consider muscle disease in children with elevated transaminase. J Am Board Fam Med. 2012;25(4):536-40. [ Links ]
16. Veropalumbo C, Del Giudice E, Esposito G, Maddaluno S, Ruggiero L, Vajro P. Aminotransferases and muscular diseases: a disregarded lesson. Case reports and review of the literature. J Paediatr Child Health. 2012;48(10):886-90. [ Links ]
Endereço para correspondência | Dirección para correspondencia | Correspondence
Joana Extreia
Rua das Vieiras, número 16, 3º andar - direito
2830- 572 - Palhais, Barreiro
E-mail: joana.extreia@gmail.com
Conflitos de interesse
As autoras declaram não ter conflitos de interesses.
Recebido em 30-12-2013
Aceite para publicação em 17-10-2014

