











 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
Introdução
A síncope define-se como uma perda de consciência abrupta e breve com resolução espontânea. Trata-se de uma condição frequente em idade pediátrica, estimando-se que até atingir a idade adulta 15% das crianças terão pelo menos um episódio de síncope.1 Esta manifestação clínica conta com múltiplas etiologias nesta faixa etária. Entre as causas mais frequentemente descritas encontram-se a síncope vaso-vagal, que corresponde a cerca de 50% dos casos, além da hipotensão ortostática ou a exposição a tóxicos. Raramente pode ser provocada por anafilaxia ou golpe de calor. Apesar de a maioria dos casos ser de etiologia benigna, as causas potencialmente fatais deverão ser obrigatoriamente excluídas. Habitualmente as causas graves de síncope são do foro cardiovascular, cursando com uma redução abrupta do débito cardíaco provocada por cardiopatia estrutural e/ou arritmia ou, menos frequentemente, bradicardia.1 Por sua vez, as taquiarritmias poderão estar relacionadas com fatores exógenos ou podem corresponder a anomalias eletrofisiológicas, como a síndroma de QT Longo. A síndroma de QT Longo tem uma prevalência estimada de 1 em cada 2.000 recém-nascidos saudáveis.2-3 Neste contexto, e perante um doente em idade pediátrica que procura os cuidados de saúde por um episódio de síncope, a história clínica e exame objetivo minuciosos, complementados com eletrocardiograma, constituem passos essenciais na correta abordagem do doente, uma vez que, embora pouco frequentes, estas causas podem ser fatais.
Descrição do caso
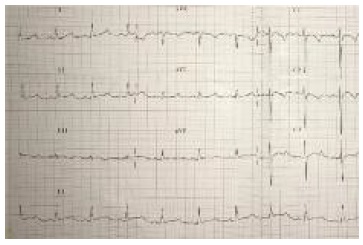
Adolescente de 12 anos, previamente saudável, observado no serviço de urgência (SU) por primeiro episódio de perda de consciência (PC), em contexto de levante súbito, com cinco minutos de duração e hipertonia generalizada. Mãe com episódios de PC classificados como epilepsia. Sem outros antecedentes familiares relevantes, nomeadamente história familiar de surdez, patologia cardíaca em idade jovem ou morte súbita. O exame objetivo não demonstrou alterações e o eletrocardiograma (ECG) revelou-se normal para a idade. Foi referenciado à consulta de neuropediatria, onde realizou eletroencefalograma e cujo resultado não evidenciou alterações. Apresentou novo episódio de PC 18 meses depois, após se ter assustado com um toque de campainha. Recuperou espontaneamente em segundos, recorrendo novamente ao SU. Nesta altura apresentava exame objetivo sem alterações, repetiu ECG, que mostrou ritmo sinusal e intervalo QT corrigido prolongado (511 ms) (Figura 1). Foi referenciado a consulta de cardiologia pediátrica, onde realizou estudo de metabolismo fosfocálcico com doseamentos normais, holter de 24 horas e prova de esforço, tendo sido diagnosticada síndroma de QT Longo. Foi observado em consulta de genética, onde efetuou estudo molecular que identificou mutação em heterozigotia dos genes KCNH e ANK2, confirmando o diagnóstico clínico. Iniciou terapêutica farmacológica com nadolol e foi fornecida ao adolescente e à família a lista de fármacos desencadeadores de taquiarritmia, encontrando-se assintomático ao longo de 12 meses de seguimento. Após revisão da história clínica materna foi pedida avaliação por cardiologia, com confirmação do mesmo diagnóstico e identificação da mesma mutação genética. No rastreio de familiares diretos foi diagnosticada também a irmã com a mesma síndroma.
Comentário
A síndroma de QT Longo é uma anomalia da repolarização miocárdica, podendo ser congénita ou adquirida. A sua forma congénita caracteriza-se por transmissão autossómica dominante (síndroma de Romano-Ward) ou recessiva (síndroma de Jervell e Lange-Nielsen), a primeira com atingimento unicamente cardíaco e a segunda associada a surdez neurossensorial, estando descritas várias formas da doença de acordo com a mutação genética identificada.4 O diagnóstico é feito com base no prolongamento do intervalo QT determinado através de ECG, correspondendo a valores superiores a 460 ms na criança pré-pubere, 470 ms no rapaz púbere e 480 ms na rapariga púbere, associado a história clínica e antecedentes familiares que apoiam a suspeita clínica.5 Contudo, esta duração é influenciada por fatores como o estado autonómico, alterações elecrolíticas, fármacos e variações circadianas. Por este motivo, é possível a obtenção de um ECG sem alterações num determinado momento num doente com síndroma de QT Longo congénito.6 Embora possa ser assintomática, esta anomalia, por cursar com episódios de síncope e resultar em arritmia ventricular, confere aumento do risco de morte súbita. Classicamente associa-se a taquicardia ventricular polimórfica ou Torsade de Pointes, tipicamente desencadeadas por fatores ambientais como ruído, exercício ou stress. Estão descritos também fármacos que propiciam o seu desenvolvimento (Tabela 1), pelo que a educação do doente e familiares, alertando para este facto, é de extrema importância.4 Perante o diagnóstico de síndroma de QT Longo congénito deverá ser realizado rastreio aos familiares diretos, com realização de ECG e estudo genético. A terapêutica passa pela administração e beta-bloquedores como nadolol, que mostrou ser mais eficaz comparativamente com outros fármacos desta classe.7-8 A dose habitual em idade pediátrica é de 1-1,5mg/Kg/dia em dose única em doentes com 12 ou mais anos e dividida em duas tomas em doentes com menos de 12 anos de idade.9
Tabela 1 Fármacos que prolongam o intervalo QT (adaptado de https://crediblemeds.org/pdftemp/pdf/CombinedList.pdf)

O risco de eventos cardíacos não é completamente eliminado pela farmacoterapia.10 Nesse sentido, é fundamental promover modificações do estilo de vida, com evicção do exercício físico extenuante, reduzir exposição a sons altos abruptos (LQTS2) e evitar todos os fármacos que podem prolongar o intervalo QT. Geralmente os beta-bloqueadores são suficientes para controlar os eventos arrítmicos, mas pode ser necessário em casos raros realizar uma desenervação simpática esquerda ou implantação de CDI. O seguimento dos doentes deve ser continuado por toda a vida, com holter anual para despiste de eventos arritmicos e medição manual do QT.10
Embora sendo um relato de caso, esta descrição traduz vários dos problemas tipicamente associados a esta patologia, como a má interpretação da etiologia de episódios de síncope, a falha no diagnóstico tanto da mãe como da criança, além da importância do rastreio familiar (neste caso, com a mãe e irmã positivas), incluindo aconselhamento genético numa futura gravidez. Assim, os autores pretendem alertar para a doença que, embora rara, pode ser facilmente detetada com um simples ECG.
Em suma, perante um doente com episódio de síncope é fundamental excluir origem cardíaca, dado que, embora menos frequentes, estas etiologias podem ter um curso fatal.

