











 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
Introdução
Em 1866, os médicos Laurence e Moon descreveram uma condição genética que se manifestava por atraso mental, baixa estatura, hipogenitalismo, ataxia, paraplegia espástica e nistagmo. Outros doentes também apresentavam retinopatia pigmentar ou atrofia coroideia. Em 1920, o médico francês Bardet e, em 1922, o patologista checo Biedl descreveram, de forma independente, um síndroma caracterizado por retinopatia pigmentar, obesidade, polidactilia, atraso mental e atrésia anal. Em 1925, os investigadores Weiss e Solis-Cohen uniram as duas entidades, designando-as de síndroma de Laurence-Moon-Biedl.1 Contudo, mais recentemente foi estabelecido que os síndromas de Laurence-Moon e Bardet-Biedl constituem patologias diferentes.
O síndroma de Bardet-Biedl (SBB) tem por base uma mutação genética. O amplo espectro clínico observado neste síndroma resulta da significativa heterogeneidade genética. A doença é transmitida quase sempre de forma autossómica recessiva, mas em alguns casos foi descrita a hereditariedade oligogénica. Até à data foram identificados mais de vinte genes diferentes como sendo responsáveis por este fenótipo. Estes genes desempenham papéis importantes nos cílios, estruturas presentes na superfície de diferentes tipos de células, intervenientes no movimento celular e em diferentes vias de sinalização química, sendo também essenciais na perceção de alguns estímulos sensoriais (visão, audição e olfato). Assim, uma alteração ao nível destes cílios resulta na alteração de todas as funções em que participam, por exemplo, a nível renal e ocular. 1-4
Estima-se que a SBB tenha uma prevalência de aproximadamente 1:150.000 pessoas, sendo que as populações com um alto nível de consanguinidade ou provenientes de regiões isoladas apresentam maior frequência da doença.
O diagnóstico da SBB é clínico e pode ser confirmado por análise molecular. 1 As manifestações principais da SBB são: distrofia da retina, obesidade, polidactilia, anomalias geniturinárias e dificuldades de aprendizagem. 1Como manifestações secundárias e menos frequentes destacam-se: estrabismo, catarata, astigmatismo, anosmia, perda de audição, sindactilia, braquidactilia, doença cardíaca congénita, situs inversus, fibrose hepática, diabetes mellitus e síndroma de Hirschsprung. 1 Os critérios para o diagnóstico da SBB incluem quatro características principais ou três características principais + duas características secundárias. 1,4
Pela natureza do acompanhamento efetuado na gravidez pelo médico de família (MF), em primeira linha de cuidados e de forma longitudinal, este desempenha um papel primordial na suspeição e orientação dos pacientes que padecem deste síndroma, bem como na referenciação precoce em gestações futuras.
Descrição do caso
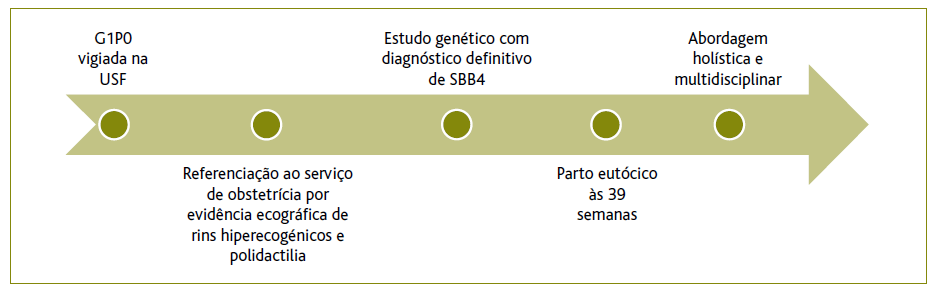
Descreve-se o caso de um utente de quatro anos de idade, do sexo masculino, raça caucasiana e natural de Lisboa. É o primeiro filho de uma fratria de três e nasceu por parto eutócico de uma gestação de termo.
A gravidez, sem fatores de risco identificados, foi vigiada na sua USF, sendo posteriormente, por alterações na ecografia morfológica - rins poliquísticos e polidactilia - referenciada ao serviço de obstetrícia da área de residência. Às 29 semanas foi referenciado à consulta de diagnóstico pré-natal da maternidade de referência.
Iniciou seguimento na consulta de genética médica no hospital pediátrico de referência por diagnóstico pré-natal (DPN) de polidactilia pós-axial bilateral das mãos e pés e rins hiperecogénicos. Foi realizado estudo genético em amostra de vilosidades coriónicas para esclarecimento da causa molecular das malformações fetais apresentadas e foram identificadas as variantes c.642+1G>T - patogénica - e c.926A>C(p.Asn309Thr) - provavelmente patogénica - em heterozigotia no gene BBS4. Foram também colhidas e estudadas amostras de sangue dos progenitores, que permitiram determinar a disposição em trans destas variantes, condicionando uma heterozigotia composta. Variantes patogénicas em heterozigotia composta no gene BBS4 são responsáveis pela SBB tipo 4, tendo assim sido estabelecido o diagnóstico definitivo.
A mãe da criança foi internada às 39 semanas para vigilância materno-fetal e programação do parto, tendo em conta a distância à maternidade do local de residência. O parto de termo, em 2018, foi eutócico e decorreu sem intercorrências. Após o parto foram realizados na maternidade o rastreio de doenças metabólicas, o rastreio auditivo e o rastreio de cardiopatias congénitas, todos sem alterações. Foi posteriormente referenciado às consultas de pediatria, genética, nefrologia, oftalmologia, otorrinolaringologia e cirurgia pediátrica plástica e de cardiologia pediátrica.
Mantém vigilância em consulta de nefrologia, onde realiza periodicamente análises e imagiologia renal. Na última ecografia renal e suprarrenal (novembro/2021) destaca-se que “ambos os rins demonstram contornos bosselados, acentuado aumento da ecogenicidade do parênquima, com má diferenciação corticomedular, traduzindo nefropatia médica. No rim direito observa-se ligeiras ectasias caliciais, o grupo calicial superior atingindo 6,7 mm de calibre, sem evidência dilatação piélica ou ureteral. No rim esquerdo visualizam-se alguns focos punctiformes hiperecogénicos condicionando cone sombra acústica, no complexo central, que relacionamos com microlitíase, sem sinais obstrutivos”.
Foi observado em consulta de oftalmologia aos seis meses de idade, da qual teve alta por não apresentar alterações naquela data. Aos nove meses foi também observado em consulta de cardiologia pediátrica, onde realizou ecocardiograma transtorácico, que revelou um coração anatómica e funcionalmente normal, tendo tido alta. No entanto, com aproximadamente um ano de idade, por relato de baixa acuidade visual, foi novamente referenciado a consulta de oftalmologia, encontrando-se a aguardar agendamento. Por iniciativa dos pais foi observado em consulta particular de oftalmologia, tendo sido diagnosticada miopia.
Aos três anos de idade foi submetido a desarticulação de dedos supranumerários da mão direita e do pé esquerdo. Mantém seguimento na consulta de cirurgia pediátrica plástica, planeando-se posterior desarticulação dos dedos supranumerários da mão esquerda e pé direito.
A criança tem mantido sempre vigilância na consulta de saúde infantil e juvenil da USF, apresentando desenvolvimento estaturo-ponderal adequado. Tem ainda regularmente consulta privada de nutrição.
Os pais mantiveram sempre adesão ao plano de seguimento e às abordagens terapêuticas propostas, com uma ótica de esperança e de expectativa em relação a eventuais medidas futuras por parte dos profissionais de saúde a cargo do caso.
O MF procurou, contudo, manter os pais informados informar acerca das possíveis complicações deste síndroma, respeitando a quantidade de informação desejada. Isto permitiu um encontro de agendas, de forma que os progenitores enquadrassem devidamente a patologia e tivessem expectativas realistas. Puderam também antecipar e planificar a resposta a eventuais exigências de cuidados por parte da criança. (Figura 1).
Comentário
Este caso clínico descreve uma doença genética que apresenta vários desafios clínicos, nomeadamente por ser rara, clinicamente heterogénea, multissistémica e de diagnóstico molecular. 1-4 Deste modo, cabe ao MF a suspeição clínica e a referenciação precoce, identificando e valorizando as alterações na ecografia morfológica (no caso descrito, os rins poliquísticos e a polidactilia) e o encaminhamento da grávida ao serviço de obstetrícia de referência, de onde será referenciada à consulta de DPN. Foi através do estudo genético, realizado para esclarecimento da causa molecular das malformações fetais encontradas, estabelecido o diagnóstico definitivo de SBB tipo 4.
Após o parto cabe ao MF participar, de forma partilhada e longitudinal, no seguimento da criança, o que pode representar um desafio desde a aquisição de conhecimentos sobre o síndroma, passando pela avaliação da criança em consulta e também à articulação e coordenação de cuidados entre as várias especialidades que possam estar envolvidas no acompanhamento da criança. Por outro lado, a incerteza proveniente do desconhecimento dos pais quanto à doença e ao plano terapêutico pode originar sentimentos de angústia e preocupação ou inclusivamente perturbações psicossociais a que o MF deve estar atento, identificando e gerindo adequadamente.
Conforme mencionado, o plano terapêutico deve ser estabelecido por uma equipa multidisciplinar, sendo fundamentalmente de suporte e não existindo um tratamento curativo para todas manifestações clínicas. Estas manifestações são abordadas de forma sobreponível à utilizada na população em geral, como é o caso das alterações visuais, da obesidade, dos problemas renais ou das dificuldades de aprendizagem. 1-4
Na visão, a retinopatia pigmentar, que ocorre em mais de 90% dos casos, leva a perda progressiva da acuidade visual, perda de visão periférica e cegueira noturna, terminando, em 75% dos casos, com a perda total da visão, por volta dos vinte anos. 5 A obesidade pode estar associada a outros problemas de saúde (dislipidemia, hipertensão arterial, diabetes mellitus, entre outros). 6 Deve salientar-se que as intercorrências nefrológicas constituem a principal causa de morbimortalidade, podendo inclusivamente determinar a necessidade de transplantação renal. 1-4 Destaca-se, sobretudo no sexo masculino, a infertilidade. 4 Já a polidactilia e algumas anomalias genitais poderão ser tratadas com cirurgia. 4 Também as alterações neuropsiquiátricas, de que se destaca a dificuldade de aprendizagem, associada a défice de atenção e processamento lento, podem merecer particular atenção. 1,4
Convirá salientar que, mesmo não existindo tratamento curativo, o diagnóstico precoce é importante para antecipar e facilitar a coordenação do acompanhamento multidisciplinar. Deste modo, recomenda-se o encaminhamento da grávida a consulta de DPN, o que permite minimizar o risco de complicações e oferecer uma melhor qualidade de vida ao doente e à família. No caso descrito, a mãe da criança foi referenciada na segunda e terceira gestações, apenas sendo confirmada SBB no terceiro filho. Evidencia-se aqui o papel do MF como principal gestor da criança e família.
Em suma, a SBB é uma doença genética multissistémica rara, cujas diversas manifestações podem surgir ao longo de todo o ciclo de vida, sendo que o MF tem um papel crucial na identificação precoce de alterações suspeitas, na sinalização clínica e em assegurar que o seguimento, muitas vezes multidisciplinar, não é descontinuado. Apresenta igualmente um papel fulcral na avaliação e eventual abordagem de problemas intercorrentes de ordem psicológica, familiar, social ou existencial.
Contributo dos autores
Conceptualização, GH e VG; metodologia, GH e VG; software, GH e VG; validação, GH e VG; análise formal, GH e VG; investigação, GH e VG; recursos, GH e VG; curadoria de dados, GH e VG; redação do draft original, GH e VG; revisão, edição e validação do texto final, GH e VG; supervisão, GH e VG. Todos os autores leram e concordaram com a versão final do manuscrito.

