











 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
Introdução
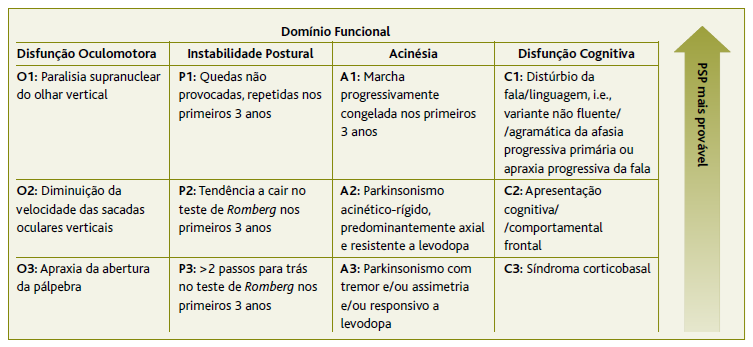
A paralisia supranuclear progressiva (PSP) é uma doença neurodegenerativa rara e esporádica, pertencente ao grupo dos síndromas parkinsónicos atípicos, que apresenta como sintomas cardinais a instabilidade postural com quedas precoces e a paralisia supranuclear vertical do olhar conjugado.1 A PSP tem uma maior incidência entre os 50-70 anos e estima-se que a sua prevalência global seja aproximadamente de 2-7 casos/100.000 indivíduos, sem predominância de género. 2 É uma doença neurodegenerativa pertencente ao grupo das taupatias, uma vez que é definida patologicamente pela agregação intracerebral de proteína tau, envolvendo caracteristicamente repetições de quatro microtúbulos, em tranças neurofibrilares, em diferentes áreas do cérebro,1 pelo que o seu diagnóstico definitivo envolve a deteção destes microtúbulos em exame anatomopatológico post mortem.1 Contudo, existem critérios de diagnóstico clínicos basilares que auxiliam na suspeita do diagnóstico e que foram recentemente atualizados pela Movement Disorder Society (MDS) de forma a tornarem-se mais sensíveis (Tabela 1). 1 Com base nestes novos critérios, os sintomas podem ser agrupados em quatro grandes domínios, que incluem a disfunção oculomotora, instabilidade postural, acinésia e disfunção cognitiva, sendo depois definido um nível de certeza de acordo com o tipo de sintoma manifestado. 1 No entanto, numa fase precoce, as manifestações da doença são na maioria dos casos inespecíficas, com alterações cognitivas e do comportamento a surgir em 30% dos casos como forma de apresentação. 3 O seu diagnóstico constitui, assim, um desafio para a comunidade médica, prevendo-se que a sua incidência e prevalência estejam subestimadas e que muitos doentes não cheguem sequer a ser diagnosticados. 4 Adicionalmente foram descritas diversas variantes clínicas da PSP, estando já identificados nove fenótipos para além do primordialmente descrito como síndroma de Richardson por Steele Richardson e Olszewski em 1964. 1,5-6 Atualmente o seu tratamento é meramente sintomático, tendo por base a melhoria da qualidade de vida destes doentes. 7
Este relato pretende descrever o caso clínico de uma mulher de 69 anos com PSP. Embora pouco prevalente, esta patologia apresenta morbilidade importante. Espera-se que a partilha de casos dentro da comunidade médica contribua para a identificação precoce de sinais de doença noutros doentes, considerando a PSP como um diagnóstico diferencial nos casos de suspeita de demência ou de depressão.
Descrição do caso
Doente do sexo feminino, caucasiana, 69 anos, reformada, casada e residente em casa própria com o marido. Inserida numa família nuclear estadio VIII do ciclo de vida familiar de Duvall e pertencente a uma classe socioeconómica média-alta (classe II), segundo a classificação de Graffar. Trata-se de uma mulher com antecedentes pessoais de hipertensão arterial e hipotiroidismo, sem antecedentes cirúrgicos e familiares de relevo, medicada com indapamida 1,5 mg e levotiroxina 0,088 mg, prescritos pelo médico de família e por endocrinologista privado, respetivamente. Sem registo de alergias medicamentosas e com o plano nacional de vacinação atualizado.
Em dezembro/2018 recorreu sozinha a consulta aberta, na sua unidade de saúde familiar (USF) e foi observada por se sentir mais triste, com choro fácil, falta de iniciativa e esquecimentos frequentes, que tinham um mês de evolução. À anamnese referia ainda episódios de ansiedade, negando conflitos familiares ou outro tipo de fator desencadeante. Negava ainda incumprimento terapêutico, distúrbio do sono ou alterações do apetite. Ao exame objetivo (EO) apresentava-se muito chorosa, orientada no tempo e no espaço, com humor depressivo, mas sem ideação suicida. Assumindo-se o diagnóstico de depressão associado a ansiedade foi iniciada sertralina 50 mg, pedido um estudo analítico com função tiroidea e agendada consulta de reavaliação em janeiro com o seu médico de família (MF).
Na consulta programada, com o seu MF, em janeiro/2019, a doente referia melhoria das queixas do humor, mas mantinha os défices de memória e negava alterações motoras ou sensitivas dos membros, cefaleias ou traumatismo recente. Ao EO apresentava-se globalmente emagrecida e exibia um discurso pobre, pouco fluente e uma lentificação psicomotora que esta não identificava como um problema. Quando questionado, o marido referia que a utente apresentava por vezes dificuldade na “expressão da fala”, interrompendo frequentemente as frases sem as terminar e acrescentava que a doente tinha receio de subir e descer escadas e também de conduzir, sem nenhuma explicação aparente para esses medos. Apresentava um mini-mental state de 28 e um exame neurológico sem outras alterações de relevo. A função tiroideia apresentava-se dentro dos parâmetros de normalidade, pelo que foi pedido um estudo analítico e uma tomografia computorizada (TC) do crânio, para exclusão de causas secundárias de um transtorno cognitivo leve. Na consulta de fevereiro, a doente levou os resultados dos exames que revelavam um défice de vitamina B12 (137 pg/ml; referência: 250-1100 pg/ml) e a TC que não mostrava alterações que justificassem o quadro clínico (nomeadamente sem sinais de acidente vascular cerebral recente), tendo sido prescrita uma suplementação com cianocobalamina. Tendo em consideração os achados foi ainda realizada a referenciação da doente aos cuidados de saúde hospitalares de neurologia (Figura 1).
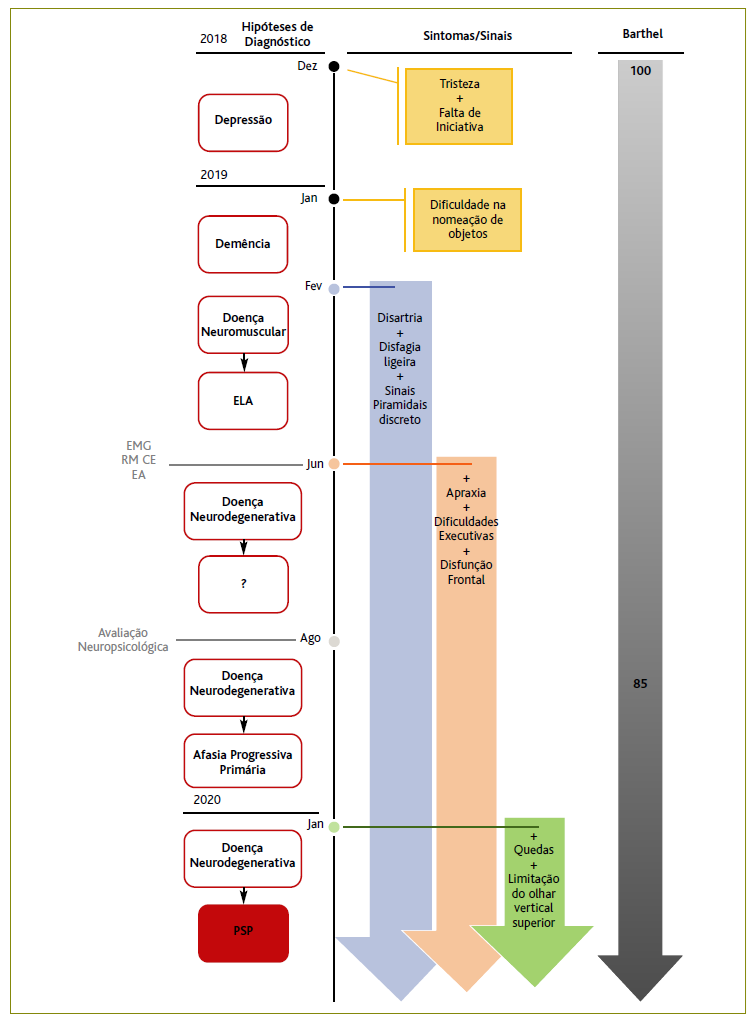
Figura 1 Esquema sumário das consultas, sintomas/sinais, hipóteses de diagnóstico, exames e escala de Barthel ao longo do tempo. Legenda: EA = Estudo analítico; ELA = Esclerose lateral amiotrófica; EMG = Eletromiografia; CE = Cerebral; PSP = Paralisia supranuclear progressiva; RM = Ressonância magnética.
Em março/2019, a doente foi observada em consulta de neurologia no hospital público onde referia um agravamento do distúrbio da fala associada a uma dificuldade na articulação das palavras. Negava disfagia, diplopia, défice motor dos membros ou alterações sensitivas. Ao EO a doente exibia disartria, reflexos osteotendinosos ligeiramente vivos, com marcha normal e sensibilidade e força muscular conservadas, sem afasia, neglet ou alterações dos campos visuais e oculomotricidade, sem dismetria e instabilidade postural/desequilíbrio. Os sinais foram interpretados em contexto de uma doença neuromuscular (DNM) provável, pelo que foram pedidos uma eletromiografia (EMG), ressonância magnética cerebral (RM CE), RM cervical, estudo analítico e videofluoroscopia. A EMG não evidenciou sinais de disfunção do 2.º neurónio, a RM CE demonstrou focos glióticos inespecíficos, sem lesão ocupante de espaço ou lesões do tronco, a RM cervical excluiu a presença de alterações medulares, a videofluoroscopia demonstrou a existência de disfagia ligeira e as análises apresentaram-se dentro dos parâmetros de normalidade. Perante os resultados excluiu-se DNM, nomeadamente esclerose lateral amiotrófica de fenótipo bulbar, tendo-se prescrito clopidogrel 75 mg e atorvastatina 20 mg para um controlo mais rigoroso dos fatores de risco cardiovasculares. Foi ainda aconselhada a fazer terapia da fala para a disartria, para reforço da força da faringe e aumento da resistência muscular da língua e agendada reavaliação em três meses.
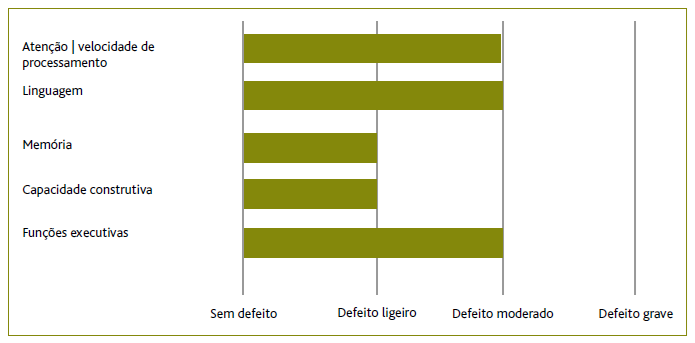
Na consulta, em junho/2019, a doente mantinha a disartria, mas acrescentava dificuldades na escrita e nas funções executivas, nomeadamente na tomada de decisão, mantendo a capacidade para fazer todas as atividades de vida diárias (AVD) (Barthel=100). Ao EO destacava-se a presença de apraxia bilateral e disfunção frontal, demonstrada por riso fácil alternado com quase choro, sem sinais extrapiramidais, quedas ou limitação do olhar, pelo que se colocou em hipótese a presença de uma doença neurodegenerativa. Neste contexto foi pedida uma avaliação neuropsicológica para caracterização do funcionamento cognitivo e para apoio no diagnóstico diferencial. Esta avaliação, realizada em agosto/2019, utilizando seis diferentes escalas psicométricas (Montreal Cognitive Assessment, Auditory Verbal Learning Test, Escala de Memória de Wechsler, teste das trilhas, teste de Stroop e Clinical Dementia Rating), evidenciou a presença de defeitos moderados na nomeação e escrita, bem como no domínico executivo e da atenção, um processamento cognitivo globalmente lentificado, um discurso não fluente, com evidente esforço articulatório e alterações na memória verbal, concordantes com a presença de um quadro de afasia progressiva primária (Figura 2).
Em janeiro/2020, numa consulta de reavaliação em neurologia, a doente evidenciou um agravamento dos sintomas prévios e descreveu a presença de quedas ocasionais, não provocadas, para os lados, e alguma limitação nas AVD, principalmente na higiene, no vestir-se e no cozinhar (Barthel=85). Ao EO destacava-se a presença de impulsividade motora, discreta limitação do olhar vertical superior e dificuldade nos movimentos oculares de perseguição que a neurologista associou ao diagnóstico de PSP e iniciou rivastigmina em baixa dose (4,6 mg/24 h). Foi enviada carta para o MF a explicar o quadro clínico, pelo que se decidiu conjuntamente pedir terapia ocupacional e reiniciar terapia da fala.
Atualmente, além das terapias e do treino de marcha que executa diariamente com o apoio do marido, a doente mantém seguimento em consulta hospitalar de neurologia e acompanhamento na sua USF. Nas consultas de acompanhamento com o MF foi determinada uma classificação de 65 pela escala de Barthel, correspondente a dependência moderada, estando dependente do marido para o banho, para se vestir e para deambular. Pela instabilidade postural foram reforçadas as medidas de prevenção de quedas e aconselhada a utilizar espessante na água para prevenção de aspiração. Foram ainda abordados os problemas de insónia e retenção urinária, que a doente apresenta atualmente, tendo sido prescrita quetiapina 25 mg e tansulosina 0,4 mg. Apesar da doença grave num elemento da família, segundo o APGAR familiar de Smilkstein, trata-se de uma família altamente funcional (score=10), contando com o apoio diário por parte dos filhos do casal. De acordo com a escala de Zarit, o marido, que é o cuidador e o representante legal, não se apresenta sobrecarregado (score=15); no entanto, é feita uma avaliação regular da família e do cuidador por parte da equipa de saúde (médico e enfermeiro de família) de forma a identificar e intervir precocemente em qualquer elemento/evento desestabilizador desta harmonia.
Comentário
Destacando o papel do MF, este é responsável pela prestação de cuidados continuados longitudinalmente ao doente, assim como pela gestão da doença que se apresenta de forma indiferenciada, numa fase precoce da sua história natural, características que colocam o MF numa posição privilegiada para diagnosticar, acompanhar, intervir e referenciar nas situações como a que é apresentada no presente caso clínico. 8
A doente em questão começou por apresentar labilidade emocional, disfunção cognitiva e distúrbio da linguagem, que são sintomas precoces da PSP. 9 No entanto, a identificação do distúrbio da linguagem não foi detetada na sua primeira consulta em 2018, tendo sido apenas percebido pelo seu MF na consulta programada. Estas alterações manifestadas pela doente podem estar presentes em diversas patologias, como demência, depressão, doença neuromuscular e doenças neurodegenerativas (Figura 2); no entanto, os doentes com disfunção cognitiva na PSP habitualmente não se queixam de défices mnésicos. Apresentam-se apáticos, desatentos, desinteressados e indiferentes ao mundo que os rodeia. 5,9 Tornam-se mais lentos a processar novas informações e perdem a capacidade de tomar iniciativas e decisões, o que se designa como uma demência do tipo subcortical. 5 Apesar de a doente ter inicialmente apresentado queixas de défice mnésico, estes sinais de demência subcortical também estavam presentes no caso descrito.
Adicionalmente, o tipo de relação de confiança que se estabelece entre MF, utente e respetiva família permitiu a exposição complementar de sinais (nomeadamente os receios de conduzir e subir/descer escadas) pelo marido da doente, valorizando o contributo da família no acompanhamento deste caso que, apesar de não terem sido valorizados inicialmente, poderiam já constituir sinais precoces da limitação vertical do olhar, ser uma instabilidade postural ou uma acinésia. A utilização de meios complementares de diagnóstico possibilitou excluir alguns diagnósticos diferenciais (DNM, doença medular e acidente vascular cerebral) e só a evolução temporal da doença, com a progressão da sintomatologia, permitiu o estabelecimento do diagnóstico definitivo. A instabilidade postural e a disfunção oculomotora, característicos desta patologia, foram essenciais neste processo de diagnóstico. De ressalvar que o tipo de quedas que estes doentes apresentam são caracteristicamente no plano sagital posterior e a limitação do olhar vertical costuma ser predominantemente no olhar inferior. 9 No entanto, nesta doente, as quedas iniciaram-se no plano coronal e a limitação do olhar foi detetada nos movimentos oculares superiores, o que não é o mais característico da patologia.
Atualmente o tratamento da PSP é meramente sintomático e paliativo e o seu prognóstico é reservado, com uma sobrevivência média após o diagnóstico de apenas três a quatro anos. 10 A disfagia constitui um problema crítico a ser abordado precocemente dado o risco de pneumonia de aspiração, que constitui a principal causa de morte nestes doentes. 7 A rivastigmina para o tratamento da disfunção cognitiva parece também mostrar resultados promissores. 11 Porém, dado o caráter debilitante e multissistémico da doença, o seguimento destes doentes deve ser feito por uma equipa multidisciplinar, no qual se inclui o MF como elo de ligação entre os diferentes profissionais de saúde e coordenador de cuidados. O MF desempenha, assim, um importante papel na suspeição diagnóstica e na referenciação atempada do doente para cuidados de saúde hospitalares mais especializados e tem ainda um papel importante na identificação de famílias/cuidador com sobrecarga e na adequação de recursos existentes na comunidade às progressivas necessidades demonstradas por parte do doente e sua família, garantindo uma melhor qualidade de vida dos doentes com PSP.

