











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
A 19-year-old male with a previous diagnosis of Ehlers- Danlos syndrome type IV (EDS), diagnosed by genetic study during adolescence due to a suggestive family history, presented to the hospital with large volume hemoptysis. He had a long-time history of sporadic cough and blood- tinged sputum. Physical examination revealed a temperature of 37ºC, a heart rate of 128 beats/min, a blood pressure of 108/52mmHg, a respiratory rate of 17breaths/min, and a pulse oximetry value of 92% in ambient air. Chest auscultation revealed decreased respiratory sounds on the right hemithorax. Laboratory tests were within normal limits, with hemoglobin of 13,8 g/L. Contrast-enhanced chest computed tomography (CT) revealed bilateral peribronchovascular ground-glass opacities (GGO) involving all the pulmonary lobes, and probable diffuse alveolar hemorrhage was reported. Large hemorrhagic cavitary lesions with thick walls and air-blood levels in the right lower lobe were also present (Fig. 1). The patient was hospitalized and treated conservatively with oxygen therapy, cough suppressive therapy (opiates) and anti-fibrinolytic therapy (aminocaproic acid). During the hospitalization the patient remained clinically stable, maintaining intermittent episodes of small-volume hemoptysis.
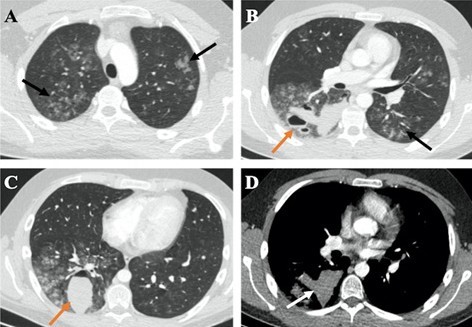
Figure 1: Late arterial phase chest CT depicts from cranial to caudal (A to C, respectively) bilateral peribronchovascular ground-glass opacities (GGOs) involving all the pulmonary lobes, especially the right lung (black arrows) and thick-walled cavitary lesions with air-blood levels in the right lower lobe (orange arrows). The high density of blood-containing cavities is illustrated in D (white arrow). The GGOs translate diffuse alveolar hemorrhage and the cavitary lesions result from the spontaneous rupture of the lung. There was no evidence of hemothorax or pneumo- thorax.
On the 7th day of hospitalization, a reevaluation contrast-enhanced chest CT was performed revealing unexpected bilateral pulmonary thromboembolism. The multifocal GGOs disappeared but the hemorrhagic cavities had enlarged (Fig. 2). There were no signs of right ventricle disfunction on echocardiogram or deep vein thrombosis in the lower limbs on Doppler ultrasound. After a multidisciplinary discussion, the patient was not considered for invasive interventional or surgical treatment due to the high risk of vascular manipulation. Also, hypocoagulation was not initiated due to the hemorrhagic risk, and also because of the clinical stability and absence of referred signs and symptoms. On the 9th day of hospitalization, the patient had an episode of syncope and died shortly after, due to right ventricle dysfunction.
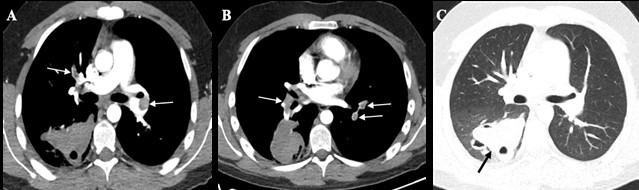
Figure 2: CT pulmonary angiogra- phic study shows bilateral pulmonary thromboembolism (arrows in A and B) and enlarged hemorrhagic cavity in the right lower lobe (arrow in C). No signs of pulmonary hypertension or right ventricular strain were present.
The estimated prevalence of EDS is between 1/10.000 and 1/25.000, among which EDS type IV is the most severe type and accounts for about 5 to 10%.1 EDS type IV, also known as the vascular type EDS, is a rare inherited autosomal dominant disease caused by a defect or deficiency in the pro?α1 chain of type III collagen encoded by the COL3A1 gene.2 The genetic disorder confers an anomalous fragility of blood vessels, uterus, hollow viscera, skin, and lung. The classic clinical manifestations are thin and hyperelastic skin, easy bruising, characteristic facial features (thin and pinched nose, prominent cheek-bones, sunken or bulging eyes, thin upper lip), and joint hypermobility.1 Additionally, patients with EDS type IV are prone to acute vascular and visceral ruptures, mainly the sigmoid colon and gravid uterus, which are rarely observed in other forms of EDS. Vascular EDS is characterized by widespread vascular changes in any location at a young age, including aneurysms, dissections, ruptures, and fistula formation. Tear or dissection of medium and large caliber arteries is responsible for the majority of deaths and reduced life expectancy of this disorder.3 Neurological complications including epilepsy, hemiparesis, and numbness are also reported and secondary to intracranial hemorrhage or stroke, microangiopathy, and central nervous system congenital malformations.1,4 Pulmonary manifestations are uncommon. Hemoptysis occurs in 25% of the patients.5 Imaging findings may depict pneumothorax, tracheobronchomegaly, saccular bronchiectasis, emphysema, pulmonary opacities, and thick-walled hemorrhagic cavities due to spontaneous lung rupture. The lung cavities may heal producing scars or thin-walled bullae. Pneumothorax may result from the spontaneous rupture of the lung parenchyma or subpleural bullae.2,4,5,6,7 Our case is distinctive because of the unusual presentation of this disorder coursing with large hemorrhagic cavitations and bilateral pulmonary thromboembolism. Usually, EDS type IV presents with hemorrhagic complications and thrombotic events are exceptionally rare. Thrombotic events may be attributed to the defect of collagen that supports blood vessels, making them more prone to injury with consequent damage of endothelial cells and activation of the coagulation cascade.8 Other factors instead of EDS, such as hypercoagulable disorders (positive antiphospholipid antibodies, factor V Leiden mutation), may also be responsible.8,9 Invasive imaging studies such as conventional angiography and other percutaneous interventions should be discussed on a case by case basis because of the risk of vascular trauma related to catheter insertion and vascular manipulation.3

