











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
Case Report
A 69-year-old Caucasian male attended the unscheduled attending service with complaints of easy fatigue and dyspnea on minor efforts, which had been evolving over three months and had recently worsened. Lately, he reported anorexia, a swollen abdomen and the development of lower limb edema.
The patient had one history of controlled hypertension, insulin-dependent type 2 diabetes mellitus, and monoclonal gammopathy of undetermined significance (MGUS) since 67 years old.
Physical examination only showed lower limb edema and an enlarged abdomen with dullness on the flanks.
An abdominopelvic computed tomography (CT), without contrast (because of the patient's atopic history to contrast), was performed which revealed bulky, partially calcified retroperitoneal adenopathy encasing the aorta and inferior vena cava and mesenteric adenopathy with the same characteristics; also, some fine and amorphous calcifications involving a few areas of the omentum and moderate ascites were detected (Figure 1A and 1B).
The diagnostic hypotheses raised were lymphoma, metastatic lymphadenopathy, Castleman disease and Amyloidosis.
The retroperitoneal adenopathy was biopsied under CT guidance, and histology confirmed the diagnosis of AL-type Amyloidosis (Figure 1C)
The patient declined the suggested chemotherapy treatment.
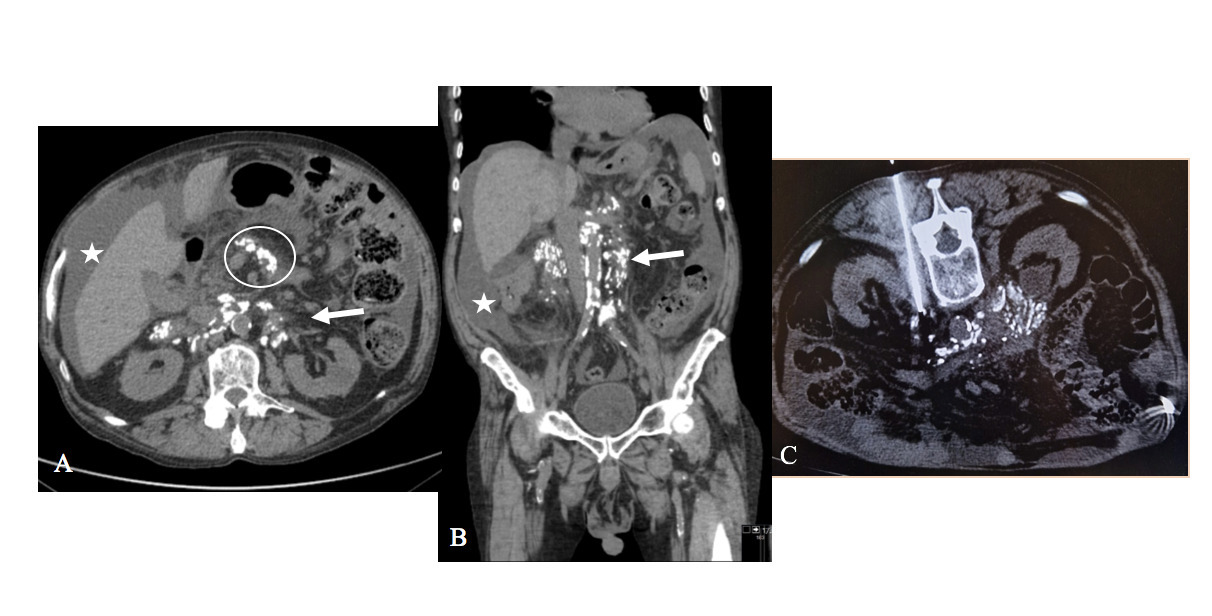
Figure 1: Amyloidosis. Images from abdominopelvic CT without contrast, axial (image A) and coronal (image B) revealing bulky, partially calcified retroperitoneal adenopathy encasing the aorta and inferior vena cava (arrow), and mesenteric adenopathy with the same characteristics (circle); also, moderate ascites is observed (star). The axial image of the biopsy under CT guidance of retroperitoneal adenopathy (image C) confirmed the diagnosis of AL-type Amyloidosis.
Discussion
Amyloidosis represents a group of rare diseases characterized by the extracellular deposition of abnormal protein fibrils, termed amyloid, in various organs and tissues, leading to organ dysfunction.2,3
Amyloidosis can be classified classically into primary, with no pre-existing disease except multiple myeloma or other plasma cell dyscrasia, or secondary, with co-existent underlying infective, inflammatory or neoplastic disease.1
Histologically, it is classified based on the chemical type that forms the fibrils, as either a type 'AA' (derived from serum protein A) or 'AL' (derived from immunoglobulin light chains).1,3
Clinically, Amyloidosis can cause localized signs and symptoms and its diagnosis relies on biopsy and histopathological examination, which reveal amyloid deposits in tissues.2
Radiology plays a role in the diagnosis by using imaging techniques and guiding tissue biopsies, as well as in the management and treatment, as it allows non-invasive visualization of amyloid deposits and associated organ damage, allowing monitoring of Amyloidosis and its response to treatments.
Ultrasound can detect associated organomegaly, mass lesions and obstruction. CT and magnetic resonance imaging (MRI) can identify and characterize amyloidosis-related organ damage. CT detects amyloid deposits, for example, in the lungs, liver, spleen and kidneys,2 and can also guide biopsies. On the other hand, MRI can detect amyloid deposits in the brain and spinal cord and identify typical features of cardiac Amyloidosis.2 In our case, brain and column MRIs were negative for amyloid deposits.
Nuclear medicine techniques (including cardiac scintigraphy and PET-CT) have provided greater specificity for the diagnosis of Amyloidosis involving the heart and central nervous system.2
Considering the result from the biopsy of retroperitoneal adenopathy, the patient's history of MGUS, and the existence of no other relevant diseases, the diagnosis of primary AL-type Amyloidosis was confirmed.
The retroperitoneum is rarely involved in patients with AL-type systemic Amyloidosis, considering the sixteen cases reported in the literature.3
Therefore, our clinical case is another rare case of retroperitoneal involvement in AL-type systemic Amyloidosis, where Radiology is essential in its diagnosis, offering several modalities to visualize amyloid deposits and guide biopsies in addition to the crucial role in managing and monitoring the progression of the disease and treatment evaluation.

