











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
Introduction
First described by James Ewing in 1921, the Ewing sarcoma family represents a group of tumours with similar morphology. This includes the Ewing sarcoma of the bone, the extraskeletal Ewing sarcoma, the peripheral primitive neuroectodermal tumour, and the small cell tumours in the chest and lung region, also known as Askin tumour.1,2
Ewing sarcoma is a malignant small round cell sarcoma that presents most commonly in the diaphysis and metaphysis of long bones of children, adolescents, and young adults.3
Extraskeletal Ewing sarcoma, first reported by Tefft in 1969, is an uncommon soft tissue tumour that shares similar morphology with the more common Ewing sarcoma of the bone.4,5,6 The former accounts for about 1% of soft tissue sarcomas, being more common in locations like the chest wall, paravertebral region and lower extremities, but other anatomical sites have been described.7
Primary pleural Ewing sarcoma is a very rare form of extraskeletal Ewing Sarcoma, with only a few cases reported in the literature.1,5,8,9,10,11 It has a high degree of malignancy, rapid growth, and a poor prognosis. Due to its rarity, it is likely to be misdiagnosed clinically.1,8
The preferred treatment includes a combination of surgery and chemotherapy.8
Case Presentation
A 20-year-old Caucasian female had a chest X-ray taken during a routine general practice appointment. The patient had no symptoms. She had no significant personal history and did not take medication regularly, apart from the contraceptive pill. The patient was a social smoker with a smoking burden of less than 3 pack years.
A posteroanterior erect chest X-ray was performed and showed a hypotransparent mass with well-defined margins localized in the middle third of the left hemithorax, about 60 mm in diameter (Figure 1 - A). It was not possible to determine its relationship with the contour of the left costal pleura. Further investigation was advised.
A contrast-enhanced thoraco-abdomino-pelvic computed tomography (CT) scan was performed and confirmed a large left pulmonary mass, non-calcified, heterogeneous, with soft tissue attenuation and some hypodense areas of water density (Figure 1 - B, C, D). The soft tissue component showed enhancement after intravenous contrast, better depicted in the iodine density map software of spectral CT, revealing an iodine density of 1.95 mg/ml in comparison with the necrotic tissue where an iodine density of 0.82 mg/ml was shown (Figure 1 - F). The lesion had no cleavage planes with the pleura and showed no signs of invasion of the adjacent bone. The protruding edge towards the lung had relatively smooth margins, indicating that the adjacent lung tissue was compressed rather than infiltrated. It measured 61 x 56 x 43 mm (longitudinal x transverse x anteroposterior axes). The CT scan showed no thoracic lymphadenopathies or distant metastasis.
For staging, a positron emission tomography (PET-CT) was performed, in which the mass presented a heterogeneous distribution of the radiopharmaceutical, with a central non-metabolic area and a peripheral metabolic halo of mild to moderate intensity (SUV of 2.3; Figure 1 - E). No other pathological deposits of radiopharmaceutical suggestive of malignancy were identified. The transthoracic percutaneous biopsy was negative for tumour cells.
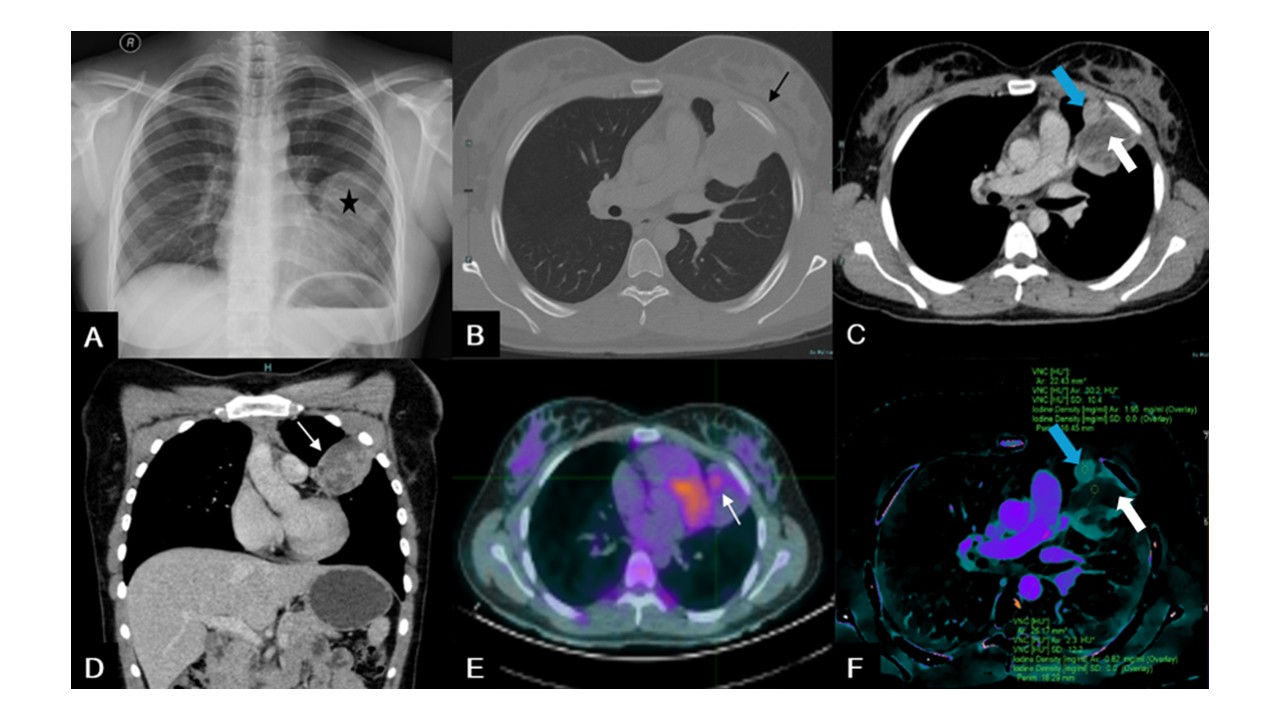
Figure 1: (A) Posteroanterior chest X-ray showing opacification of the middle third of the left hemithorax, by a mass with a well-defined superior margin (black star). Relationship with the contour of the left costal pleura is unclear. (B) Bone window axial chest CT showing a non-calcified lesion with no clear signs of invasion of the adjacent bone (black arrow). (C) Soft tissue window axial venous phase CT revealing an heterogeneous mass with soft tissue attenuation (blue arrow) and central hypodense areas of water density (white arrow). (D) Soft tissue window coronal venous phase chest CT showing the heterogeneous mass (white arrow). (E) Axial chest PET-CT scan depicting a peripheral metabolic site of the lesion with mild to moderate intensity, with a SUV of 2.3 (white arrow). (F) Axial iodine density map software of spectral CT after iv contrast showing an heterogeneous lesion with central areas of water density (white arrow) and peripheral areas of soft tissue density (blue arrow).
The patient was promptly scheduled for surgery and underwent left upper lobectomy (Figure 2 - A). Gross examination revealed a partly solid, lobulated, whitish-gray tumour, with extensive areas of haemorrhagic degeneration within the pleural thickness (Figure 2 - B). Histologically, the tumour consisted of a monomorphic population of small, round cells with round to oval vesicular nuclei and a thin clear cytoplasmic rim. Areas of necrosis, apoptosis, and mitoses were observed (Figure 3 - A, B). Immunohistochemistry revealed positive staining for Vimentin, CD99, FLI-1 and ERG, with focal expression of pS100 and synaptophysin (weak staining); the tumour cells were negative for AE1/AE3, EMA, Chromogranin A and CD56; the Ki67 proliferation index was approximately 45% (Figure 3 - C, D, E, F). Genetic analysis by fluorescent in situ hybridization (FISH) detected a rearrangement in 80% of the nuclei for the FUS gene, confirming the diagnosis of Ewing sarcoma. No EWSR1 gene rearrangements were found. Lymph node assessment showed no evidence of metastasis.
The post-operative period was unremarkable. The patient was referred for a Tertiary Centre for therapeutic decision. At multidisciplinary meeting it was decided to initiate chemotherapy with vincristin, doxorubicin, and cyclophosphamide, followed by ifosfamide and etoposide (VAC-IE regimen). The patient has completed chemotherapy, and follow-up CT scan, performed one year after surgery, showed no signs of recurrence.
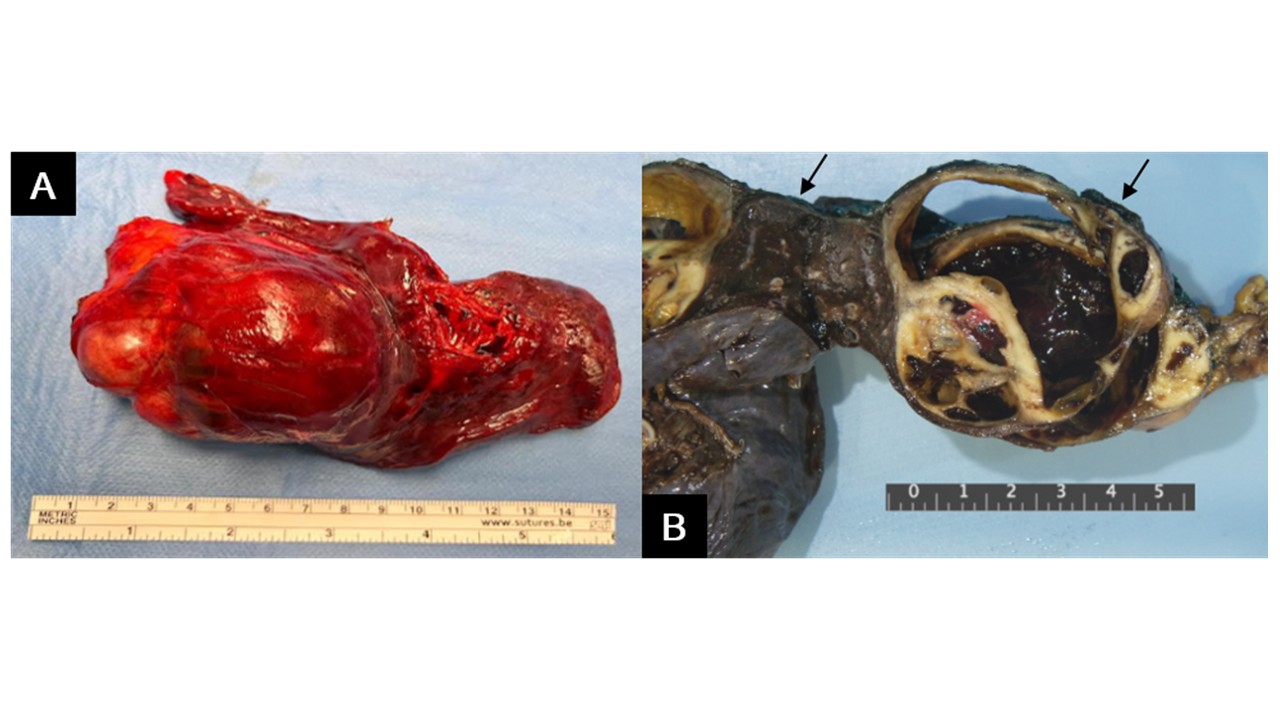
Figure 2: (A) Surgical specimen. (B) Pulmonary lobectomy specimen including a flap of parietal pleura (black arrows). A well-circumscribed tumour (6 × 6 × 5.5 cm) is located within the pleural thickness, causing adhesion between the pleural layers. The tumour is solid, lobulated and whitish-gray, with extensive haemorrhagic degeneration. It compresses but does not invade the pulmonary parenchyma.
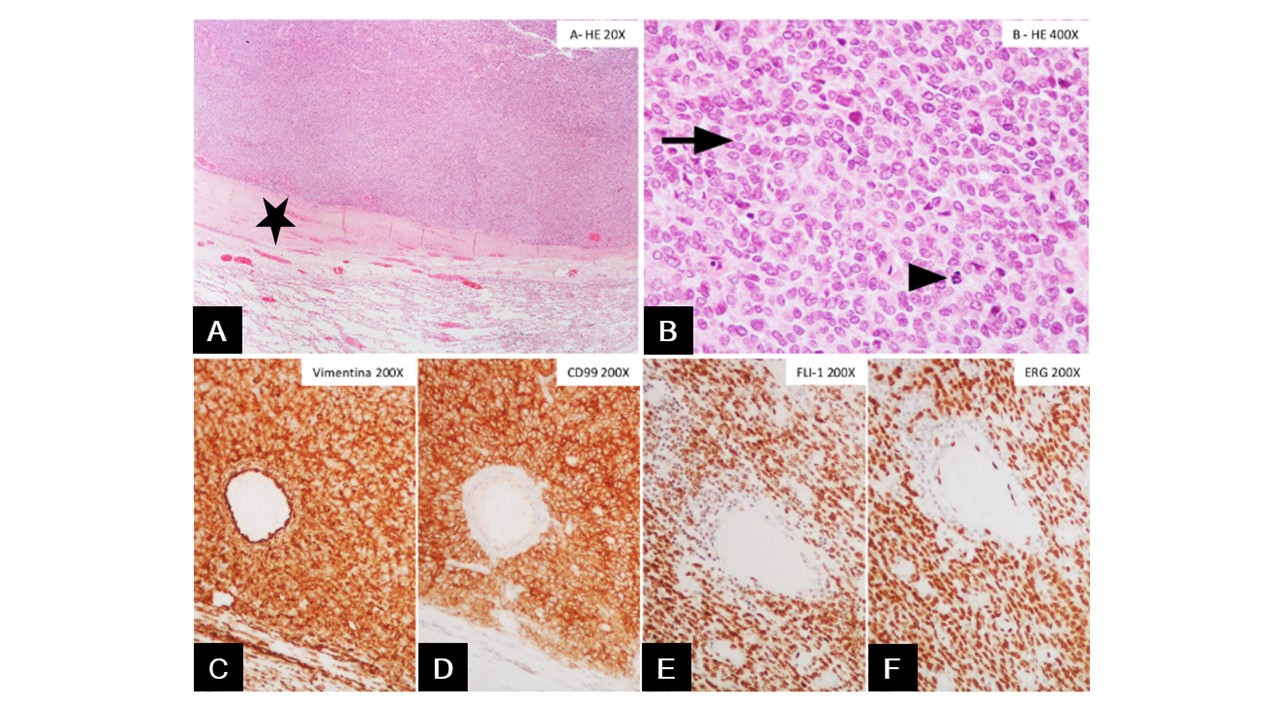
Figure 3: (A) Tumour with a sheet-like growth pattern, well circumscribed and separated from the pulmonary parenchyma by the thickened pleura (black star). (B) Tumour cells are uniform small round cells, with round nuclei, finely stippled chromatin, inconspicuous nucleoli, and scant clear to eosinophilic cytoplasm with indistinct cytoplasmic membranes. Occasional mitotic figures are observed (arrows). (C and D) Immunohistochemistry shows strong, diffuse membranous expression of Vimentin and CD99. (E and F) Nuclear staining is present for both FLI1 and ERG.
Discussion
We present a well-documented case of a twenty-year-old female patient who was diagnosed with primary pleural Ewing sarcoma.
This particular extraosseous Ewing sarcoma is an extremely rare entity, with only sporadic cases described. It has a bad prognosis and its diagnosis is challenging since it has no specific clinical manifestations or characteristic imagiological features. Therefore, the final diagnosis is based on histopathology, immunohistochemistry and genetic analysis of the tumour specimen.11,12
The most frequent clinical manifestations are dyspnoea, fever, chest or back pain, and cough.1 Our patient was asymptomatic. Computed tomography typically shows a large, sharply delineated mass and the post-contrast enhancement is usually intense and heterogeneous, with hypodense foci, often resulting from intralesional necrosis. These findings are non-specific and the differential diagnosis includes rhabdomyosarcoma, malignant fibrous histiocytoma and liposarcoma.12
For that reason, the definitive diagnosis is achieved by CT-guided core-needle biopsy or pathological examination of the resected specimen. In our case, the transthoracic percutaneous biopsy was negative for tumour cells, but as the clinical suspicion for neoplasm was high, the patient was promptly referred for surgery in a highly differentiated Centre. Diagnosis was made by histological, immunohistochemical and genetic analysis of the surgical specimen.12
The treatment of choice for localized disease is surgical resection. Chemotherapy can precede or follow the surgical procedure, and its regimens include vincristine + dactinomycin + cyclophosphamide + doxorubicin or vincristine + dactinomycin + cyclophosphamide. Ifosfamide and etoposide can be subsequently added, although there are no significant differences in survival rates. Radiotherapy is performed in unresectable localized tumours.11
Conclusion
The diagnosis of primary pleural Ewing sarcoma is based in immunohistochemistry, histopathology and FISH analysis, since clinical findings and radiological features are nonspecific.
This case underlines the importance of maintaining a high index of suspicion for large thoracic masses in young patients. The presence of well-defined margins and a wide pleural base in imaging exams may provide a hint for the final diagnosis.

