











 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
Introdução
O primeiro caso de duplicação intestinal foi descrito por Calder em 1733, mas só em 1941 é que Ladd e Gross definiram esta patologia como se tratando de “estruturas ocas, esféricas ou alongadas, com parede de músculo liso, revestidas por mucosa, intimamente ligadas a alguma porção do tubo digestivo.”1
A incidência desta patologia é de 1:5000 nados-vivos, sendo mais frequente no sexo masculino. Geralmente são lesões únicas, podendo haver lesões síncronas em 10%-20% dos casos. São diagnosticadas antes dos 2 anos em 80% dos doentes.2-4
A localização mais comum é o ílion (30%) e válvula ileocecal (30%), seguidos do jejuno (8%), cólon (6%-7%) e reto (5%). A duplicação pode ser de 3 tipos, segundo McPherson: tipo I, quistos simples; tipo II, diverticular; tipo III, tubulares. O seu diagnóstico baseia-se nos critérios de Rowling: 1 - duplicação da parede em continuidade com a do órgão duplicado; 2 - quisto coberto por
parede muscular lisa; 3 - presença de mucosa do trato digestivo, podendo existir por vezes mucosa heterotópica.5,6
Têm surgido várias propostas para tentar explicar a origem desta patologia, nomeadamente a teoria de Bremer (1944) que sugere ser secundária a erros de recanalização do tubo digestivo durante a fase embrionária.7
As duplicações completas são raras. As formas quísticas, mais frequentes, geralmente não comunicam com o lúmen; as tubulares podem ter uma ou mais comunicações.
As duplicações do trato digestivo são em 13% dos casos de origem colorretal e devem ser abordadas cirurgicamente quando encontradas para evitar futuras complicações.8
O quadro sintomático pode apresentar-se por obstipação, perfuração, hemorragia digestiva ou oclusão intestinal. A estratégia cirúrgica requer uma ressecção conjunta da área duplicada até pelo menos 2 cm além da duplicação. A cirurgia pode ser dificultada por fibrose na área de duplicação, presença de neoplasias e alterações no suprimento sanguíneo.4,8
As duplicações tubulares do cólon com extensão abaixo da reflexão peritoneal podem estar associadas a malformações do aparelho genito-urinário.9
Caso clínico
Apresentamos o caso de uma doente de 60 anos de idade, com antecedentes pessoais conhecidos de hipertensão arterial e lúpus eritematoso sistémico.
A doente recorreu ao serviço de urgência por queixas de obstipação e dor abdominal com vários dias de evolução. Ao exame objetivo apresentava um abdómen distendido, doloroso à palpação nos quadrantes inferiores, com massa palpável no hipogastro. Ao toque retal apresentava a ampola colapsada por compressão extrínseca.
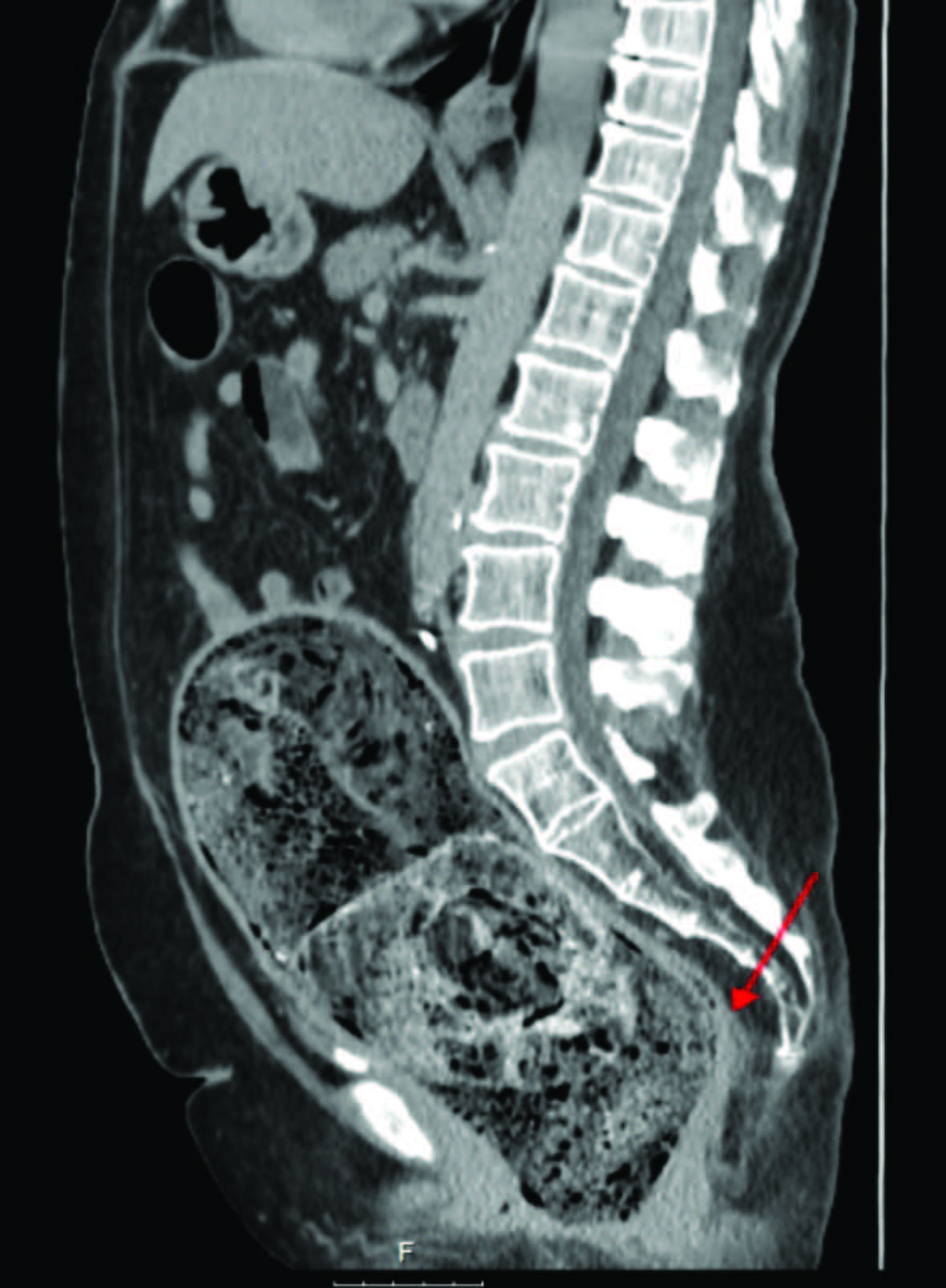
FIGURA 1: TC mostrando fecaloma condicionando compressão do lúmen funcional apontado pela seta a vermelho.
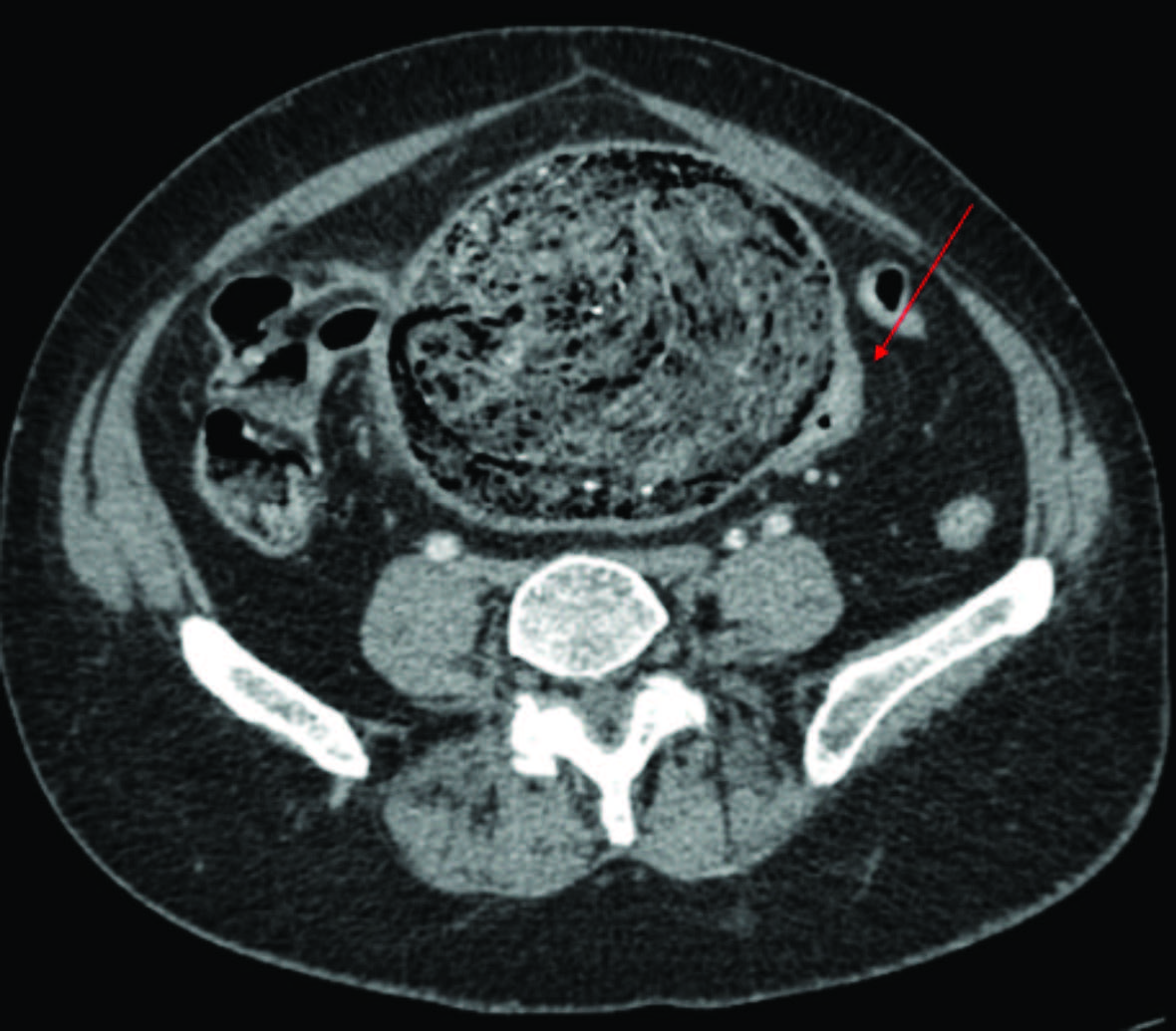
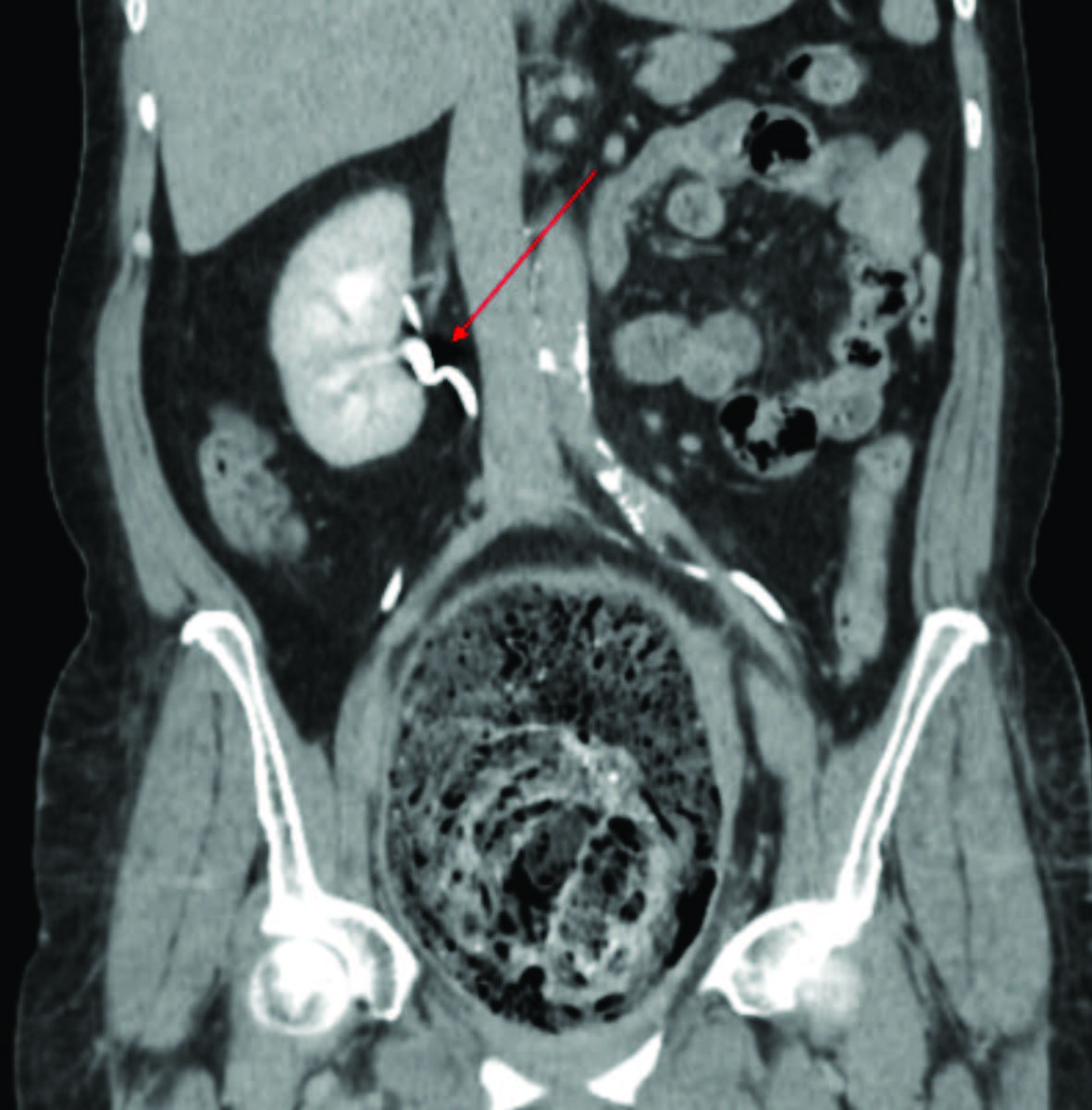
Figuras 2 e 3: Imagens de TC demonstrando oclusão por volumoso fecaloma da transição retossigmoideia, com lúmen funcional colapsado (seta na Fig. 2, em cima). Na Fig. 3, em baixo, mostra-se duplicação pielo-calicial à direita com 2 ureteres a emergirem separadamente do rim (seta vermelha).
Foram pedidos vários exames complementares de diagnóstico realizados na urgência. O estudo analítico não apresentava alterações. O radiograma de abdómen mostrava alguns níveis hidroaéreos e ausência de ar na ampola retal. Realizou tomografia computorizada (TC) abdominopélvica que mostrava “(…) volumoso fecaloma retal, medindo 23x9 cm, condicionando oclusão intestinal e espessamento parietal do cólon. Útero atrófico, desviado para a direita (…), malformação renal com rim direito resultante da fusão dos dois rins, com evidência de 2 ureteres que têm implantação vesical separada e bilateral” (Fig.s 1,2 e 3).
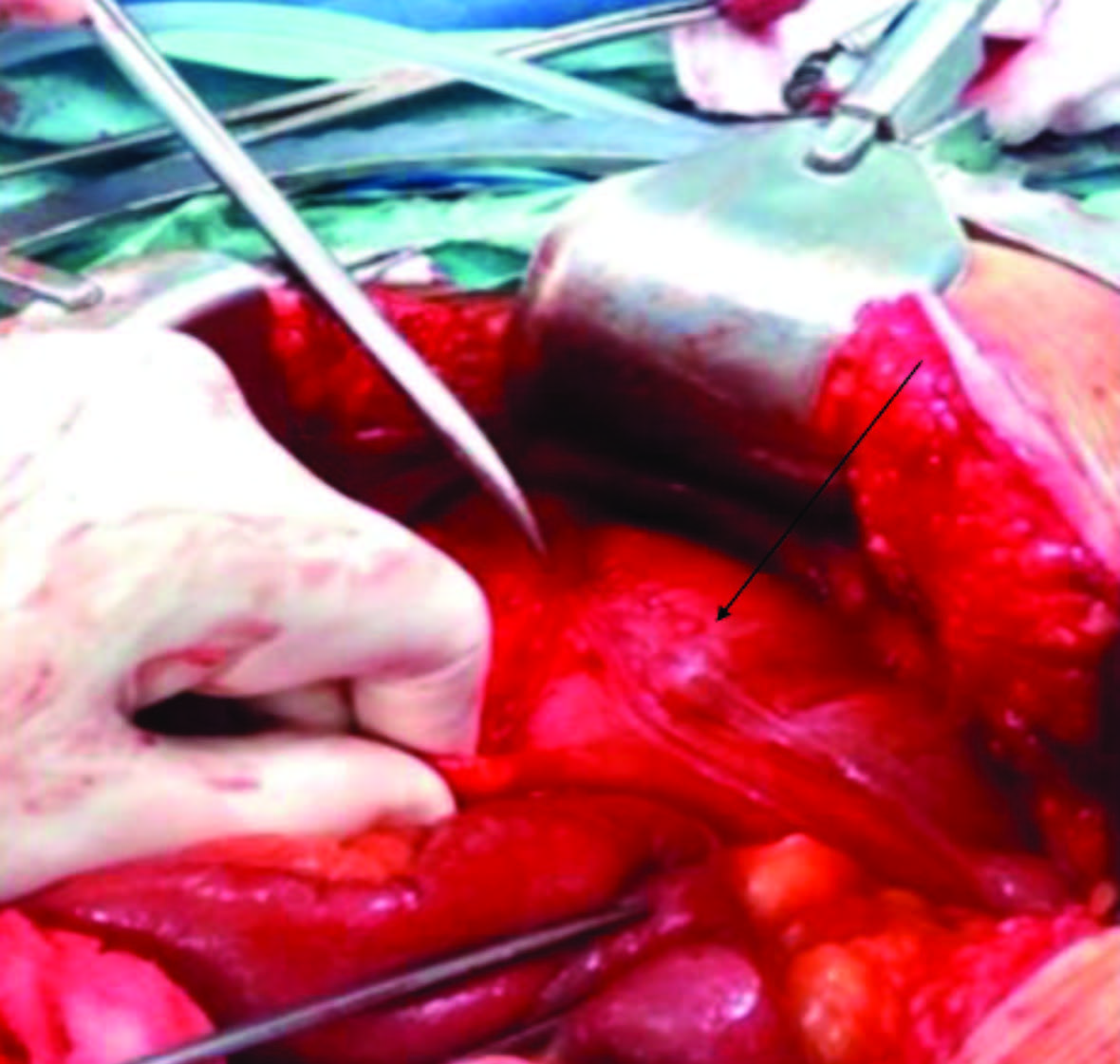
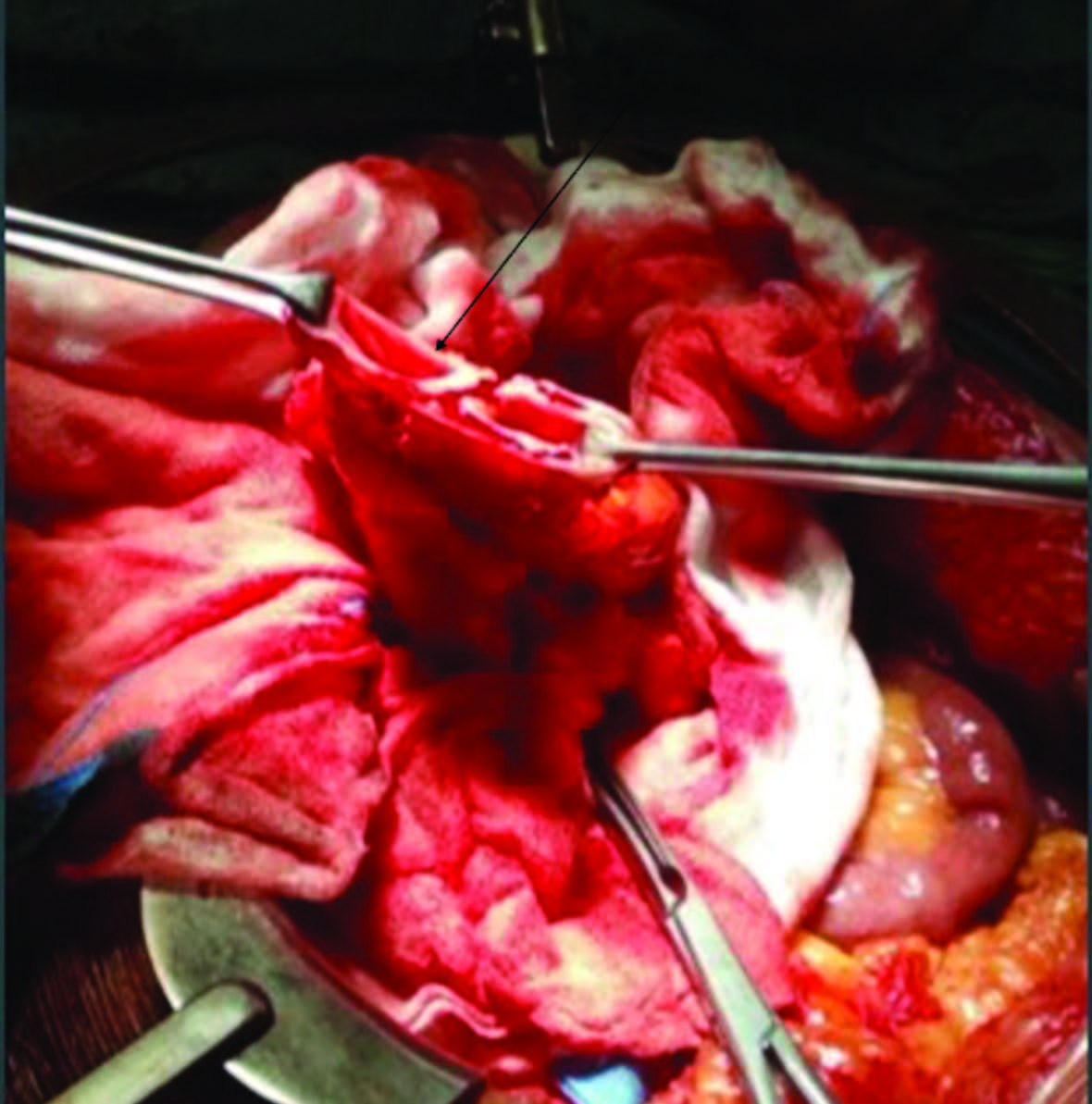
Figuras 4 e 5: Fotografia intraoperatória mostrando duplicação tubular desde o cólon sigmoide ao reto médio. A taenia coli duplicada estendia-se até ao cólon ascendente, como mostra a seta, na figura em cima. Colostomia em cano de espingarda com lúmen duplicado apontado pela seta, figura em baixo.
Foi proposta para cirurgia urgente. No intraoperatório constatou-se a presença de duplicação do cólon sigmoide e reto que terminava em fundo cego no reto médio, preenchida por um volumoso fecaloma. A taneia coli duplicada estendia-se de forma contínua até ao cego, pelo que não era possível avaliar de forma segura a extensão proximal da duplicação (Fig. 4). Optou-se por realizar uma ressecção tipo Hartmann com secção distal ao nível do reto inferior e exteriorização de ambos os lúmenes em cano de espingarda (Fig. 5).
No pós-operatório, a doente foi submetida a colonoscopia para estudo do restante cólon. O limite proximal do lúmen duplicado situava-se cerca de 10 cm proximalmente à colostomia, não sendo encontradas outras alterações no restante cólon.
Aos 6 meses foi submetida a cirurgia de ressecção da restante área duplicada e restabelecimento do trânsito intestinal, que decorreu sem intercorrências.
O exame histopatológico de ambas as peças operatórias, confirmou tratar-se de uma duplicação tubular, sem mucosa heterotópica ou neoplásica.
Discussão/conclusão
A apresentação clínica dos casos de duplicação cólica depende da sua localização, extensão, e de outros fatores, como a presença de mucosa heterotópica. A obstipação, perfuração e a oclusão intestinal são as apresentações mais comuns, atendendo a presença de um lúmen distalmente cego.
A abordagem cirúrgica depende da localização e do tipo de duplicação. As duplicações quísticas, quando pequenas, são excisadas em bloco com o cólon normal adjacente à semelhança das duplicações tubulares. Se a duplicação se estende abaixo da reflexão peritoneal devem ser excluídas malformações do aparelho génito-urinário.
A ressecção cirúrgica deve estender-se até cerca de 2 cm além do óstio do lúmen duplicado atendendo à presença de alterações fibróticas, ao suprimento sanguíneo partilhado e ao risco de desenvolvimento de neoplasia no segmento duplicado. O adenocarcinoma é o tipo de neoplasia mais frequentemente encontrado, seguido do carcinoma escamoso e do carcinoide.10

