











 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
Introdução
O tumor fibroso solitário (TFS) é um tumor raro de tecidos moles, de origem mesenquimatosa, descrito originalmente na pleura, e cujo diagnóstico e tratamento são desafiantes. Partilha aspetos histológicos com outros tumores de tecidos moles, mas os progressos no diagnóstico molecular e imuno-histoquímico têm possibilitado a sua diferenciação, sendo o CD34, STAT6 e Bcl-2 os marcadores mais importantes.1 A localização mais frequente destes tumores é intratorácica, sendo a pleura o principal, seguido do pulmão, mediastino e diafragma.2 No entanto, existem também TFS em localização extratorácica, nomeadamente no fígado, na glândula suprarrenal, na pele e na região da cabeça e pescoço,3 cujo primeiro caso foi descrito em 1991.4 Os mais comuns ocorrem na cavidade oral, mas também foram descritos na órbita, nariz e seios perinasais, nasofaringe e espaço parafaríngeo, laringe, glândulas salivares major e tiroide.3
A ocorrência de TFS no canal auditivo externo é extremamente rara e apenas existem três casos de TFS convencional reportados na literatura,5-7 existindo também um caso de TFS rico em células gigantes.8
Caso clínico
Uma doente do género feminino, de 27 anos, sem antecedentes pessoais de relevo, recorreu à consulta de Otorrinolaringologia do Centro Hospitalar Universitário Lisboa Central (CHULC) por sensação de diminuição da acuidade auditiva e vários episódios de otorreia homolateral e sensação de obstrução progressiva do canal auditivo externo esquerdo com mais de um ano de evolução. Ao exame objetivo, observou-se uma massa arredondada, de superfície lisa, revestida por pele íntegra, com cerca de 2 cm de maior eixo, localizada na parede posterior do canal auditivo externo esquerdo, causando a sua completa obstrução e não possibilitando a visualização da membrana timpânica. Realizou um audiograma tonal que revelou uma hipoacusia de condução de grau ligeiro que se relacionou com a obstrução do canal auditivo externo. A tomografia computorizada (TC) de ouvidos revelou uma massa relativamente homogénea, hipodensa, sem invasão das estruturas adjacentes, ocupando todo o canal auditivo externo esquerdo, sem quaisquer alterações do ouvido médio (Fig. 1). A ressonância magnética de ouvidos confirmou a existência de uma formação tecidular obliterativa do conduto auditivo externo esquerdo, com cerca de 21 mm de diâmetro ântero-posterior, de contornos regulares e hipointensa em T1 e T2 (Fig. 2).
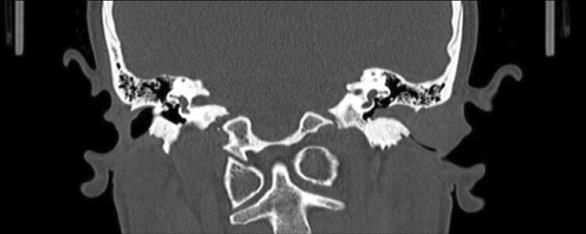
Figura 1: TC de ouvidos (corte coronal) revelando a lesão (asterisco), ocupando todo o canal auditivo externo esquerdo.
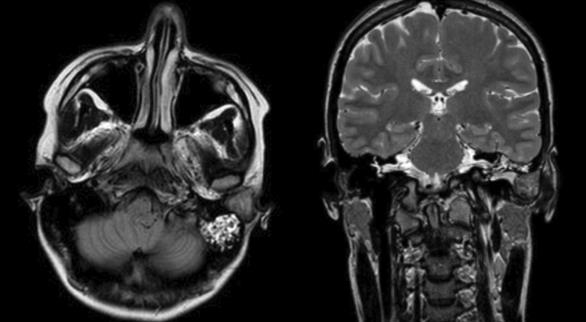
Figura 2: Ressonância magnética de ouvidos demonstrando lesão obliterativa do conduto auditivo externo esquerdo, em T2 (corte axial - seta branca, corte coronal - asterisco).
Foi realizada excisão total macroscópica do tumor, sob anestesia geral, não tendo sido registada nenhuma intercorrência. O período pós-operatório imediato decorreu igualmente sem complicações.
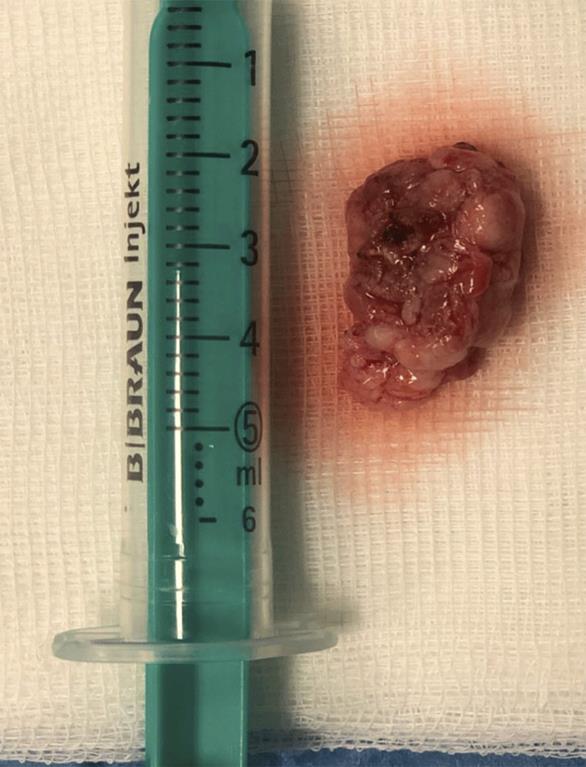
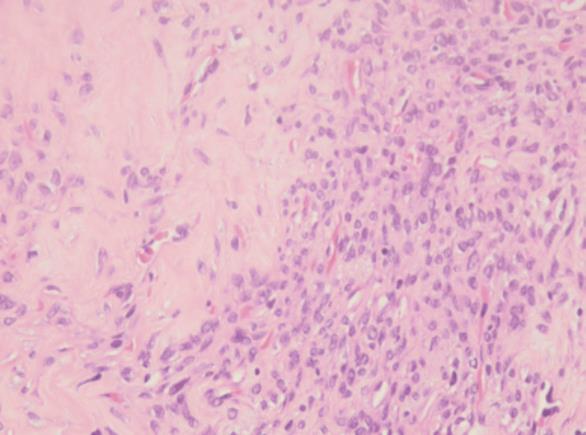
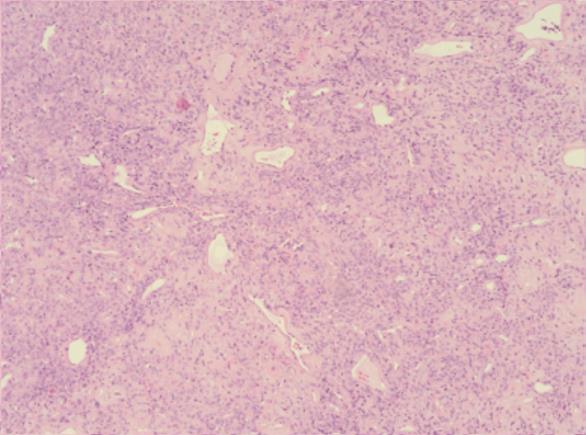
Figura 4 e 5: Proliferação de células fusocelulares e ovoides, de citoplasma escasso e núcleo alongado, sem atipia celular, dispostas num estroma colagenoso em bandas, ricamente vascularizado e com esclerose perivascular, com zonas de hipo e hipercelularidade, sem necrose. (Ampliação 100x e 400x, respetivamente)
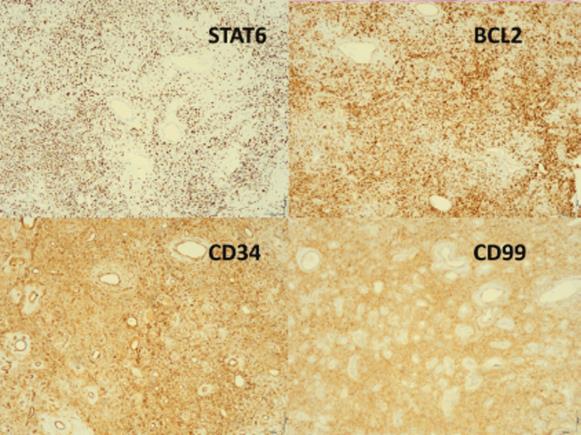
A peça cirúrgica consistia em dois fragmentos irregulares branco-acinzentados e elásticos, o maior com 2,5x1,7x1,2 cm (Fig. 3) e o menor com 0,8 cm de maior eixo. Microscopicamente, a lesão era constituída por células fusiformes ou ovais, com escasso citoplasma e núcleo alongado, por vezes com nucléolo. As células dispunham-se num estroma rico em colagénio, organizado predominantemente em bandas e rico em vasos com esclerose perivascular evidente, alternando áreas de hiper e hipocelularidade (Fig.s 4 e 5). Não existia evidência de atipia celular e o índice mitótico era baixo. O estudo imuno-histoquímico revelou reação positiva das células tumorais para o marcador CD34, vimentina, CD99, anticorpo BCl2 e STAT6 (Fig. 6), e reação negativa para a proteína S-100, cromogranina, sinaptofisina, desmina, actina do músculo liso (AML), MelanA e HMB45. Baseado nestes achados, a lesão foi diagnosticada como tumor fibroso solitário benigno. Focalmente, a neoplasia era rasante à margem cirúrgica.
No período pós-operatório houve resolução da sintomatologia, sendo que a doente manteve consultas de seguimento para vigilância, não havendo sinais de recidiva aos 6 meses de pós-operatório. Foi definida a necessidade de vigilância a longo prazo, ou seja, trimestralmente no primeiro ano, semestralmente no segundo e terceiros anos e posteriormente seguimento anual.
Discussão
Na literatura apenas existem três casos de TFS convencional no canal auditivo externo reportados na literatura,5-7 sendo que neste artigo reportamos o quarto caso.
A sintomatologia reportada no caso apresentado, nomeadamente a sensação de diminuição da acuidade auditiva e de plenitude auricular, vai ao encontro daquilo que é descrito na literatura, uma vez que os sintomas dos TFS da cabeça e pescoço são inespecíficos e estão relacionados com a presença da massa de tecidos moles na área afetada.9 O estudo de imagem da lesão apresentada está também de acordo com o descrito em relatos anteriores, pois mostra uma lesão tecidual de contornos regulares, e, particularmente na ressonância magnética, hipointensidade nas ponderações T1 e T2, o que se correlaciona com a sua natureza fibrosa.6
Embora insuficiente para o diagnóstico, o aspeto macroscópico da lesão, firme, de superfície lisa, bem circunscrita e acinzentada, está de acordo com o reportado em TFS do canal auditivo externo.5-7
Muitas vezes este tipo de tumor é erroneamente diagnosticado como outros tumores mais comuns da cabeça e pescoço, como o angiofibroma, no entanto apresenta características histológicas e imuno-histoquímicas diferenciadoras.7 As características microscópicas levam à suspeição do diagnóstico de TFS, em particular pela observação de células fusiformes ou ovais, dispostas num padrão aleatório, com alternância de áreas constituídas essencialmente por colagénio e áreas hipercelulares,10 mas não são suficientes por si só para diferenciar o TFS de outros tumores benignos de tecidos moles, como perineuromas, lipomas de células fusiformes, fibromatose desmoide e sarcomas de células fusiformes.10,11 Antigamente o TFS era classificado como hemangiopericitoma, mas pelo facto do primeiro ter uma origem fibroblástica e não pericítica, estas são hoje classificadas como diferentes entidades. A presença de áreas hipocelulares preenchidas de forma densa por colagénio e a imunorreatividade ao CD34 distinguem o TFS do hemangiopericitoma.9 Assim, o estudo imuno-histoquímico permite-nos fazer o diagnóstico definitivo de TFS, sendo que este apresenta forte imunorreatividade para o CD34, que é tradicionalmente o marcador mais consistente; e geralmente também expressa reatividade para CD99, Bcl-2 e vimentina. A maioria dos TFS não expressa reatividade para desmina, queratinas, proteína S100, CD31, catenina beta nuclear, AML e antigénio de membrana epitelial (EMA).5,11 No entanto, o marcador imuno-histoquímico mais sensível e específico para o TFS é o STAT-6.11 No caso reportado, a positividade para este marcador, em conjunto com o restante estudo, permitiu confirmar o diagnóstico e distinguir de outras entidades.
A presença de alto índice mitótico (> 4 por 10 campos de maior ampliação), zonas de necrose tumoral, hipercelularidade, pleomorfismo nuclear e alta relação núcleo-citoplasma, podem apontar para malignidade.10 Neste caso tratava-se de um TFS benigno, não existindo sinais de malignidade.
Qualquer TFS deve ser ressecado completamente sempre que possível, uma vez que, embora a maioria sejam benignos e de crescimento lento, foram descritos TFS malignos pleurais e extrapleurais. A taxa de recidiva é muito baixa após a ressecção cirúrgica completa, embora seja ligeiramente superior para TFS extratorácicos.11 No caso reportado, foi definida a manutenção de vigilância da doente a longo prazo, uma vez que não há registos suficientes de TFS do CAE que permitam excluir potencial maligno,11 apesar de não haver características suspeitas no caso reportado.
Conclusão
O TFS do canal auditivo externo é uma entidade extremamente rara. Pretendemos com a descrição deste caso alertar para a necessidade de considerar este tumor no diagnóstico diferencial de tumores de células fusiformes do canal auditivo externo. Para o seu diagnóstico, o papel da imuno-histoquímica é fundamental. É também mandatória a vigilância após terapêutica, uma vez que pouco se conhece acerca do comportamento desta entidade a longo prazo.

