











 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
Introdução
A leucemia define-se como uma proliferação neoplásica descontrolada de células hematopoiéticas na medula óssea, que posteriormente invadem o sangue periférico e outros tecidos.1,2
A leucemia linfoide crónica (LLC) é a leucemia mais comum nos países ocidentais, manifestando-se em média aos 70 anos de idade, com uma grande variabilidade clínica. É extremamente rara em crianças, sendo mais comum no sexo masculino, e tendo um curso indolente.3,4 Quando presente em mulheres parece ter um curso mais agressivo.3 A sua etiologia não é conhecida, mas pensa-se que os fatores genéticos5 e ambientais (como exposição a diversos químicos, radiação e tabaco) possam ter um papel importante, estando descrita a duplicação de risco de desenvolvimento de doença em familiares de primeiro grau de indivíduos com LLC.3 O diagnóstico estabelece-se através do hemograma, esfregaço de sangue periférico, imunofenotipagem de linfócitos B, com expressão de antigénio CD5, assim como outros marcadores típicos de células B. Estas células proliferam na sua forma madura, mas disfuncional, acumulando-se no sangue, medula óssea, nódulos linfáticos e baço.3,4 O diagnóstico definitivo requer a presença de ≥5000/μL linfócitos B no sangue periférico pelo menos durante 3 meses. A clonalidade dos linfócitos B necessita de ser confirmada por citometria de fluxo. Apenas os pacientes com doença ativa e sintomática ou com estádios avançados específicos requerem terapêutica,4 no entanto o início precoce versus tardio da terapêutica parece não ter impacto significativo nas taxas de sobrevivência. As terapêuticas disponíveis de momento, não permitem a cura da doença, com exceção do transplante alogénico de células hematopoiéticas. A esperança média de vida atual é de 10 anos (podendo variar entre 2 e >20 anos). São fatores de bom prognóstico: mutação da região variável da cadeia pesada da imunoglobulina, deleção 13q, pouca expressão de ZAP-70 e baixos níveis de CD38 na citometria de fluxo. São fatores de mau prognóstico: tempo de duplicação de linfócitos inferior a 12 meses, deleções 17p e 11q, linfadenopatias em múltiplas cadeias linfáticas, hepatoesplenomegália, anemia e trombocitopenia.4
Caso clínico
Utente do sexo masculino, 68 anos, caucasiano, com antecedentes de dislipidemia e hipertensão arterial medicada com atenolol 100 mg, bem como leucemia linfoide crónica de células B CD38-, estadio A/0 (nas classificações Binet/Rai) - baixo risco, diagnosticada há cerca de 2 anos, com seguimento em consulta de Hematologia, sem qualquer intervenção terapêutica anterior ou atual. Na imunofenotipagem de sangue periférico identificou-se população patológica de linfócitos B, monoclonal, Kapa +, de intensidade de fluorescência débil, em 97% de todos os linfócitos B da amostra, representando globalmente 55% da celularidade total. A população patológica expressa CD19+, CD20+ heterogéneo, CD5+ homogéneo débil, CD23+ homogéneo, CD79b-, CD38-, CD11c-/+ débil, CD25-/+, CD103-, CD10-, CD200+ homogéneo forte, CD43 débil, IgM-, CD31+ débil, CD22-+ (67%) débil, CD27+homogéneo, CD45+. A nível familiar destaca-se mãe com cancro cutâneo que não sabe especificar.
Sem alergias medicamentosas. Foi solicitada observação por dermatologia por quadro de lesões papulares em forma de cúpula no couro cabeludo e face, com 6 meses de evolução, de resto assintomáticas. À observação destacam-se lesões arredondadas, eritemato-rosadas, brilhantes, bem delimitadas, com diâmetros entre 1-6 mm, e de consistência duro-elástica. Bom estado geral, sem adenopatias periféricas, sem organomegálias intra-abdominais e sem lesões suspeitas noutras localizações. Dermatoscopia com vasos atípicos e glóbulos rosados dispersos nas lesões supramencionadas.
Realizou biópsia das lesões cutâneas, com o seguinte resultado histológico: “Expansão da derme e do tecido celular subcutâneo por população linfoide em toalha, com predomínio de célula pequena com algumas células linfoides de tamanho intermédio, que focalmente permeiam alguns anexos cutâneos, mas não ulcera a epiderme. Raras figuras mitóticas e de apoptose. Estudo imunohistoquímico: população linfoide com expressão de CD20 e CD5, na ausência de expressão cyclina D1. Expressão CD3 em linfócitos T locais. Compatível com envolvimento do processo linfoproliferativo com predomínio de células pequenas, de tipo LLC (leucemia linfoide crónica)”, globalmente denominado de leucemia cutis (Fig. 1).
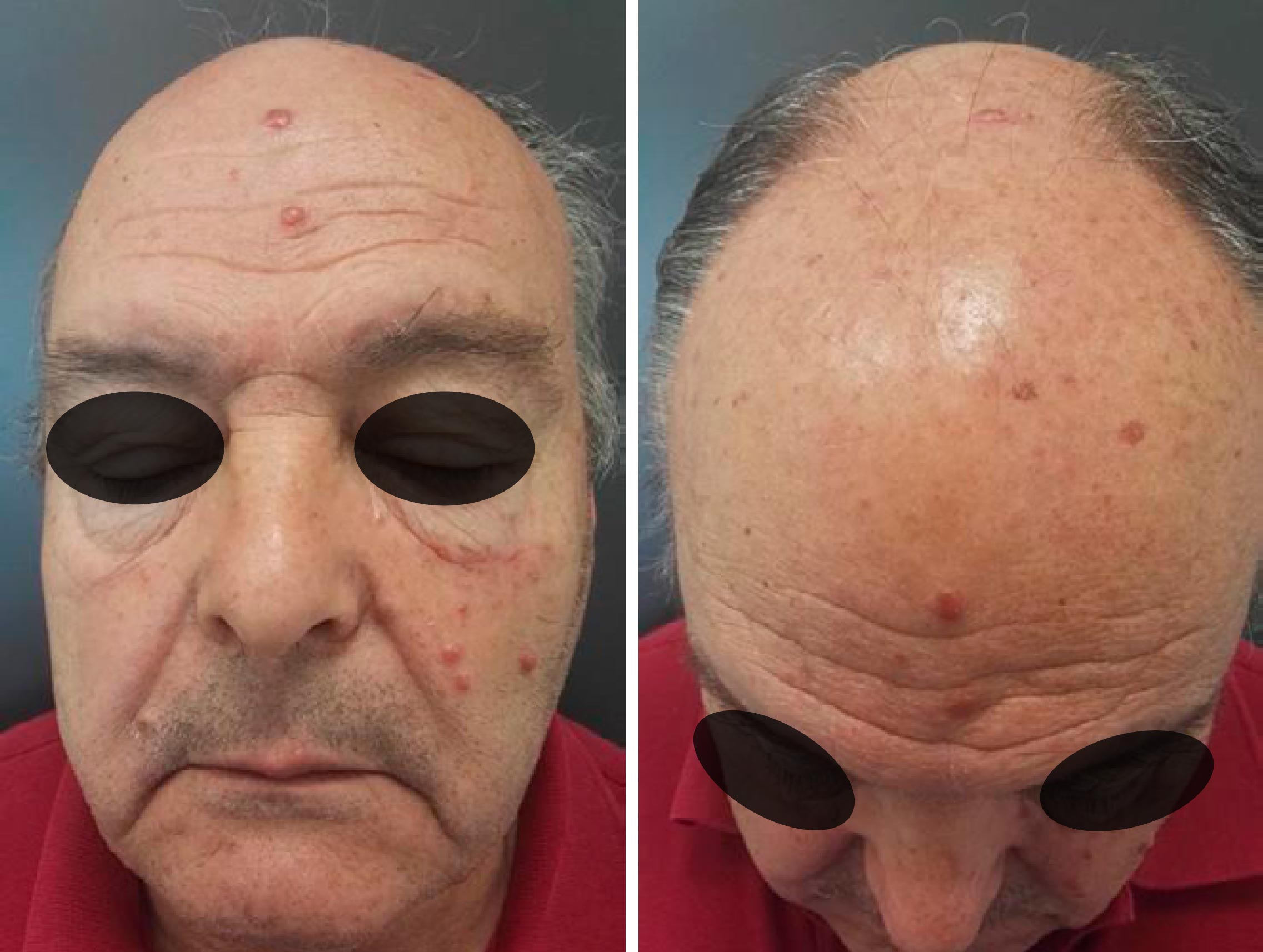
Após consulta de Hematologia com resultados analíticos dentro do esperado, optou por não realizar qualquer tipo de tratamento.
Discussão
As manifestações dermatológicas das leucemias podem ser específicas e inespecíficas. As inespecíficas são observadas com maior frequência, estando presentes em mais de 40% dos pacientes com leucemia, e resultam principalmente da manifestação das citopenias e reação a drogas, destacando-se as púrpuras, petéquias e equimoses, o prurido generalizado, eritrodermia exfoliativa, pioderma gangrenoso, síndrome de sweet, eritema multiforme, urticária, hiperpigmentação, erupções morbiliformes não específicas, e infeções oportunísticas como as micoses cutâneas, herpes zoster e úlceras crónicas secundárias a herpes simplex.6-8
As manifestações específicas resultam da infiltração de leucócitos neoplásicos no tecido cutâneo denominando-se leucemia cutis (LC), sendo de especial interesse o seu diagnóstico por ter implicações prognósticas importantes (mortalidade de ~80% um ano após o diagnóstico).1,6 A sua prevalência varia entre 3%-30%, em função do tipo de leucemia a que está associada, sendo mais frequente em idade pediátrica.1,7 O mecanismo exato pelo qual ocorre a migração de células neoplásicas para a pele não é conhecido, apesar de existiram algumas teorias.9
Existem diversos subtipos de leucemia envolvendo a pele, nomeadamente as doenças mieloides/monocíticas e as patologias linfoproliferativas (leucemias/ linfomas de células B e leucemias/ linfomas de células T).9
Apesar da LLC ser a leucemia mais comum a nível mundial, a LC é mais frequente na leucemia mieloide aguda (LMA), principalmente nos subtipos com componente monocítico.2 Na prática clínica, os casos de LMA e LLC representam respetivamente, 13% e 8% do total de casos de LC.9 Na maioria dos casos, é impossível deduzir qual o tipo de leucemia subjacente a partir das lesões cutâneas que os pacientes apresentam.10
Na idade pediátrica, a leucemia linfoblástica aguda/linfoma linfoblástico (LLA/LBL) é a neoplasia mais comum, constituindo um quarto de todos os cancros nesta faixa etária. Apesar disso, o tipo de leucemia mais associado a LC é a LMA (representando 74% dos casos, versus os 16% da LLA).8 Crianças com leucemia congénita desenvolvem LC em 25%-30% dos casos, e nestas circunstâncias, a presença desta invasão cutânea não parece representar um pior prognóstico.7-9
Nos pacientes com síndrome mielodisplásico, o envolvimento da pele pode ser a primeira manifestação de uma transformação em leucemia.2
A LC pode apresentar-se de várias formas clínicas, desde máculas, pápulas e nódulos, a úlceras e placas.
Podem ainda ser localizadas ou disseminadas, únicas ou múltiplas, e podem afetar qualquer parte do corpo.2,9 As lesões mais comuns bem como as localizações mais frequentes variam de acordo com a literatura, porém as mais reportadas são as pápulas, máculas e nódulos no tronco, cabeça e extremidades, podendo ter coloração violácea ou castanha-avermelhada ou aspeto hemorrágico.2,6-9 Quando as lesões ocorrem na face, a transição de eritema para estádio de placa ou nódulo, pode dar origem a fácies leonina, por espessamento da pele do couro cabeludo, das zonas malares e das sobrancelhas.6,8
O diagnóstico pode ser feito durante (~30%), após (mais comum) ou antes do diagnóstico hematológico (~10% - denominando-se leucemia cutis aleucémica).
Quando existe clínica cutânea, habitualmente existe concomitantemente o envolvimento de outras estruturas extramedulares (especialmente a nível do sistema nervoso central). Este diagnóstico representa assim, não só um estádio de doença avançada, como está associado a um mau prognóstico, tornando imperativa a investigação de envolvimento extramedular adicional.1,2,7,9
O facto de ter numerosas manifestações clínicas torna os resultados anatomopatológicos e imunohistoquímicos essenciais para o diagnóstico.2,10
Histologicamente, a biópsia demonstra infiltrados nodulares ou difusos na derme e/ou no tecido subcutâneo, apesar das leucemias de células T poderem ter algum epidermotropismo.9
Quando associada a LLC, a maioria dos pacientes estão assintomáticos ao diagnóstico, apresentando habitualmente linfadenopatias. No entanto, aproximadamente 10% podem apresentar-se com sintomas B, e 20%-50% com hepatoesplenomegália.11
À observação destas lesões, podemos pensar em múltiplos diagnósticos diferenciais como é o caso das metástases, linfomas, sarcoma de Kaposi, erupções secundárias a drogas, exantemas virais, sífilis, pitiríase rósea, vasculite, epidermólise bolhosa autoimune, eritrodermia idiopática, lúpus eritematoso e fibromatose gengival idiopática.6
O tratamento da LC é o tratamento da doença de base,2,10 normalmente através da quimioterapia e terapêuticas locais.7
Em suma, é importante ter em conta o diagnóstico de LC, por ser uma manifestação sistémica de mau prognóstico e agressividade, com pacientes em risco de recaída mesmo após esquemas de quimioterapia intensivos, tendo a sua maioria uma esperança média de vida inferior a um ano.6,8

