










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkGE-Portuguese Journal of Gastroenterology
versão impressa ISSN 2341-4545
GE Port J Gastroenterol vol.25 no.4 Lisboa ago. 2018
https://doi.org/10.1159/000484612
CLINICAL CASE STUDY
A Complex Case of Cholestasis in a Patient with ABCB4 and ABCB11 Mutations
Um caso complexo de colestase num doente com mutações dos genes ABCB4 e ABCB11
Mariana Ferreira Cardoso, Joana Carvalho e Branco, Vera Anapaz, Catarina Graça Rodrigues, Rita Carvalho, David Horta, Alexandra Martins, Jorge Reis
Gastroenterology Department, Hospital Professor Doutor Fernando Fonseca, Amadora, Portugal
* Corresponding author.
ABSTRACT
The low-phospholipid-associated cholelithiasis (LPAC) syndrome is a form of symptomatic cholelithiasis occurring in young adults, characterized by recurrence of symptoms after cholecystectomy and presence of hepatolithiasis. The case refers to a healthy 39-year-old Caucasian male who presented with abdominal pain and jaundice. His blood tests showed conjugated hyperbilirubinemia and elevated liver enzymes (total bilirubin 6.65 mg/dL, γ-glutamyltransferase 699 IU/L) and abdominal computed tomography revealed dilation of common bile duct and left intrahepatic ducts. Magnetic resonance cholangiopancreatography identified choledocholithiasis, retrieved by endoscopic retrograde cholangiopancreatography, after which there was a worsening of jaundice (total bilirubin 23 mg/dL), which persisted for several weeks, possibly due to ciprofloxacin toxicity. After an extensive workup including liver biopsy, the identification of two foci of hepatolithiasis on reevaluation abdominal ultrasound raised the hypothesis of LPAC syndrome and the patient was started on ursodeoxycholic acid, with remarkable improvement. Genetic testing identified the mutation c.1954A>G (p.Arg652Gly) in ABCB4 gene (homozygous) and c.1331T>C (p.Val444Ala) in ABCB11 gene (heterozygous). In conclusion, we describe the unique case of an adult male with choledocholithiasis, hepatolithiasis, and persistent conjugated hyperbilirubinemia after retrieval of stones, fulfilling the criteria for LPAC syndrome and with possible superimposed drug-induced liver injury, in whom ABCB4 and ABCB11 mutations were found, both of which had not been previously described in association with LPAC.
Keywords: Low-phospholipid-associated cholelithiasis, MDR3, BSEP, Drug-induced liver injury
RESUMO
A síndrome low-phospholipid-associated cholelithiasis (LPAC) é uma forma de coledocolitíase sintomática que ocorre em adultos jovens e que se caracteriza pela recorrência de sintomas após a colecistectomia e pela presença de hepatolitíase. O caso refere-se a um homem caucasiano de 39 anos, saudável, que se apresentou com um quadro de dor abdominal e icterícia. A avaliação laboratorial mostrou hiperbilirrubinémia conjugada e um padrão citocolestático (bilirrubina total 6,65 mg/dL; GGT 699 UI/L) e a tomografia computorizada abdominal revelou dilatação da via biliar principal e vias biliares intra-hepáticas à esquerda. A colangiopancreatografia por ressonância magnética identificou coledocolitíase, extraída por colangiopancreatografia retrógrada endoscópica, tendo-se verificado posterior agravamento da icterícia (bilirrubina total 23 mg/dL) que persistiu por várias semanas, possivelmente por hepatotoxicidade associada à toma de ciprofloxacina. Após uma extensa investigação, incluindo a realização de biópsia hepática, foram identificados dois focos de hepatolitíase em ecografia abdominal de reavaliação, pelo que se colocou a hipótese de LPAC e o doente iniciou terapêutica com ácido ursodesoxicólico, com excelente resposta. O estudo genético identificou a mutação c.1954A>G (p.Arg652Gli) no gene ABCB4 em homozigotia e a mutação c.1331T>C (p.Val444Ala) no gene ABCB11 em heterozigotia. Em suma, descrevemos o caso único de um homem com coledocolitíase, hepatolitíase e hiperbilirribinémia conjugada persistente mesmo após extracção de cálculos, cumprindo critérios de LPAC e possivelmente com sobreposição de lesão hepática induzida por fármacos, tendo-se documentado mutações nos genes ABCB4 e ABCB11, cuja associação com LPAC não havia sido previamente descrita.
Palavras-Chave: Low-phospholipid-associated cholelithiasis, MDR3, BSEP, Lesão hepática induzida por fármacos
Introduction
(LPAC) syndrome is a peculiar form of symptomatic and recurring cholelithiasis occurring in young adults, associated with mutations in the ABCB4 gene and low biliary phospholipid concentration. First described in 2001 by Rosmorduc et al. [1], it was later defined as a clinical syndrome characterized by at least two of the following criteria [2]: age at onset of biliary symptoms below 40 years, intrahepatic echogenic foci or microlithiasis and recurrence of biliary symptoms after cholecystectomy. Also, most patients report a history of cholesterol gallstones amongst their first-degree relatives [2]. Although the prevalence of LPAC syndrome remains unknown, it has been estimated to account for approximately 5% of cases of symptomatic cholelithiasis [3, 4]. The recommended medical therapy is ursodeoxycholic acid (UDCA) [5]. In the rare cases progressing to end-stage liver disease [2, 6], a liver transplant may be indicated.
We present the case of an adult male with prolonged jaundice and features consistent with LPAC, in whom both ABCB4 and ABCB11 mutations were detected.
Clinical Case
We present the case of a 39-year-old Caucasian male, with no relevant medical history, who was admitted in June 2014 for abdominal pain in the periumbilical area associated with nausea, with 2 weeks duration. In the last 48 h, the patient also noted jaundice and dark urine, with no accompanying changes in his stool. His alcohol consumption was low (5 units/week). He reported use of protein supplements 1 month before symptom onset, but denied recent use of any drugs, teas, or other herbal products. Family history included gallstone disease in his sister.
On physical examination, he was afebrile and his eyes and skin were markedly jaundiced. His abdominal examination was unremarkable and he showed no stigmata of chronic liver disease.
Laboratory workup included a hemoglobin of 16.0 g/dL, with normal white blood cell and platelet count. He had elevated liver tests: aspartate aminotransferase (AST) 170, alanine aminotransferase (ALT) 570, alkaline phosphatase (ALP) 186, and γ-glutamyltransferase (GGT) 699 IU/L; total and direct bilirubin were 6.65 and 4.95 mg/dL, respectively. There were no changes in kidney function, electrolytes, and coagulation, and C-reactive protein (CRP) was 0.3 mg/dL.
The patient underwent an abdominal ultrasound followed by contrast-enhanced computed tomography (Fig. 1), which together showed dilated left intrahepatic ducts and common bile duct, reaching 7 mm, with no obstructive lesion; no gallstones were detected. Magnetic resonance cholangiopancreatography (MRCP) identified a 4-mm stone in the proximal common bile duct, with upstream dilation (Fig. 2). Due to a progressive rise in CRP (5.4 mg/dL), the patient was put on ciprofloxacin assuming cholangitis. An endoscopic retrograde cholangiopancreatography (ERCP) was then performed, with sphincterotomy, extraction of the common bile duct stone with a basket and multiple other smaller stones, which were yellow in color. Considering the high risk of stone migration, a plastic biliary stent was placed to prevent complications prior to cholecystectomy.
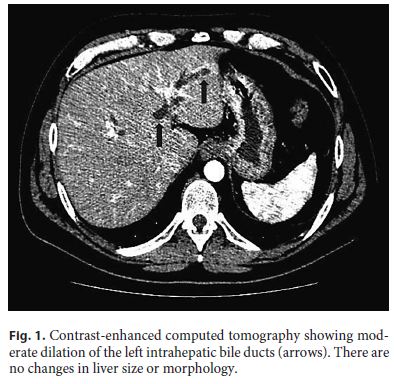
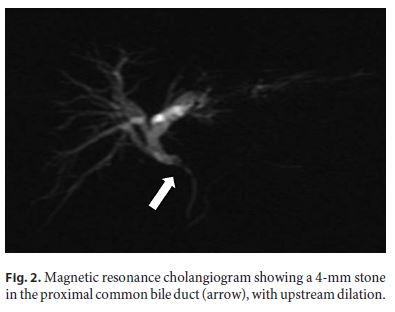
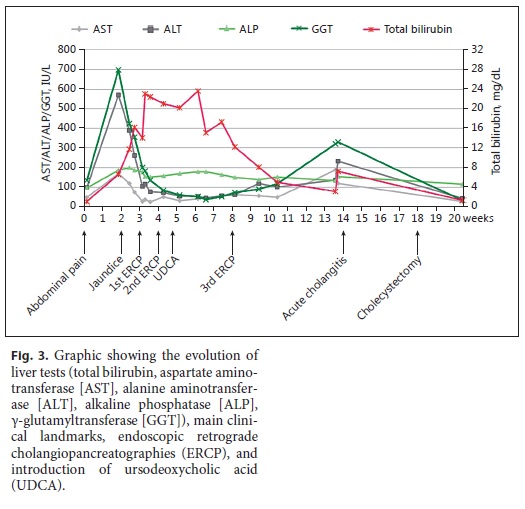
Despite a progressive decrease in AST, ALT, ALP, and GGT, only 24 h after the ERCP there was a marked rise in direct (16 mg/ dL) and total bilirubin (23 mg/dL), which remained elevated for several weeks, even after repeated ERCP with removal of additional microlithiases with a basket (Fig. 3).
An extensive workup was carried out, with exclusion of viral hepatitis (hepatitis A, B, and C, Epstein-Barr virus, and cytomegalovirus, as well as human immunodeficiency virus), autoimmune liver disease (negative antinuclear, anti-smooth muscle, antimitochondrial, anti-LKM-1, anti-neutrophil cytoplasmic, anti-gp210 and anti-Sp100 antibodies, and normal IgG4 level), and metabolic diseases including Wilsons disease (normal serum and urinary copper and a slightly diminished ceruloplasmin; no Kayser-Fleischer rings), hemochromatosis (elevated ferritin 1,109 ng/mL and transferrin saturation 60%, with a normal HFE genotyping), α1-antitrypsin deficiency, and porphyria (normal levels of urinary and fecal porphyrines and urinary porphobilinogen; normal porphobilinogen deaminase activity).
Due to the protracted jaundice with no identifiable cause, a liver biopsy was performed. Histology showed hepatic tissue with preserved architecture and scarce mixed intralobular and sinusoidal infiltrates, with predominance of mononuclear cells. There was focal centrolobular hepatocyte ballooning, focal hepatocellular necrosis (no piecemeal necrosis), and focal regenerative hepatocellular changes resembling rosettes. Mild parenchymal cholestasis and hepatocyte siderosis were observed. There was marked deposition of ceroid pigment in Kupffer cells. No significant fibrosis was present. Liver iron content was elevated (2,181 mg/g) and copper was within normal range (20.5 mg/g). These aspects were consistent with an acute hepatocellular lesion, with toxic and viral etiologies being more likely.
At this point, the patient was markedly jaundiced, had mild tenderness over the right upper quadrant, and complained of nausea, choluria, and pruritus. He showed no signs of hepatic encephalopathy and his coagulation remained normal. Eosinophil count was also normal. Having excluded other causes of liver injury, toxic hepatitis was considered to be the most likely explanation for maintained jaundice. Drugs used since hospital admission included ciprofloxacin, meropenem (both used to treat cholangitis), and propofol (used in ERCP). The iodinated contrast agent iobitridol was also used during ERCP.
Repeated abdominal ultrasound identified two foci of hepatolithiasis in segment II, the largest measuring 11 mm, with upstream dilation of the left intrahepatic ducts, with no underlying stricture (Fig. 4). Although these had not been identified in MRCP on presentation, we note that left intrahepatic dilation was already visible then.
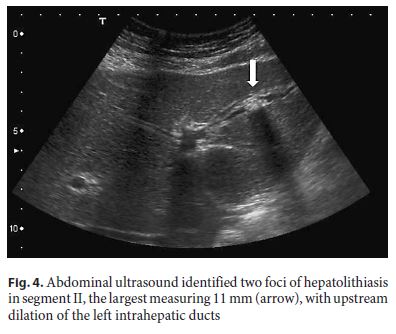
Considering age below 40 years and hepatolithiasis, as well as family history of gallstones in a first-degree relative, the diagnosis of LPAC was assumed and the patient was put on UDCA, with progressive resolution of symptoms and near normalization of liver enzymes in the following 2 months, allowing for removal of the biliary stent. Six weeks after stent removal, there was a new episode of abdominal pain due to acute cholangitis (with total bilirubin 7 mg/dL, CRP 14 mg/dL, and slight common bile duct dilation on ultrasound), with good response to treatment with ceftriaxone. He was submitted to laparoscopic cholecystectomy 1 month later.
The genetic study of ABCB4 and ABCB11 genes identified the mutation c.1954A>G (p.Arg652Gly) in ABCB4 (homozygous) and the mutation c.1331T>C (p.Val444Ala) in ABCB11 (heterozygous).
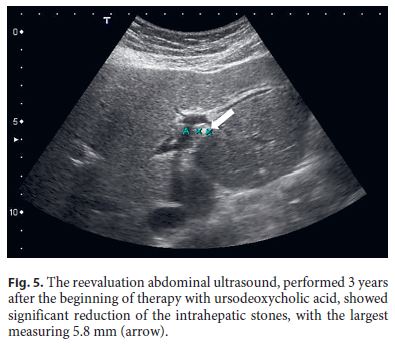
After 3 years of follow-up, the patient is still treated with UDCA and maintains surveillance with blood tests and abdominal ultrasound every 6 months. He remains asymptomatic, has normal liver tests, and his last ultrasound showed significant reduction of the intrahepatic stones (Fig. 5).
Discussion
The ABCB4 gene encodes a P-glycoprotein (MDR3: multidrug resistance 3) with flippase activity, responsible for the translocation of phosphatidylcholine across the canalicular membrane of the hepatocyte [7]. The proposed mechanism in LPAC syndrome is that a defect in ABCB4 function reduces the phospholipid content of bile and increases the cholesterol saturation [1, 2], thereby increasing its lithogenicity. The reduced formation of mixed micelles also results in bile with high concentration of free bile salts and hence high detergent properties, leading to bile duct membrane injury and cholestasis with increased serum GGT levels [2, 5].
ABCB4 defects are implicated in a wide spectrum of cholestatic diseases besides LPAC [8], including progressive familial intrahepatic cholestasis type 3 (PFIC3) [9], benign recurrent intrahepatic cholestasis type 3 (BRIC3) [10], intrahepatic cholestasis of pregnancy (ICP) [11, 12], drug-induced cholestasis [13], transient neonatal cholestasis [8, 12], and adult idiopathic liver fibrosis or cirrhosis [12, 14]. Although the relation between genotype and phenotype is not always clear, patients with homozygous or compound heterozygous mutations, particularly those leading to truncated proteins, tend to have more severe disease [8]. Indeed, patients with biallelic ABCB4 mutations resulting in absence of protein function die within the first decade if not transplanted [12], whereas patients with milder mutations may be asymptomatic until a trigger such as pregnancy, systemic infection, or a xenobiotic reduces MDR3 function below a critical threshold [8, 13, 15].
Although LPAC syndrome was originally described in association with ABCB4 mutations, in a study with 156 patients who fulfilled the diagnostic criteria, a genetic variant was only found in half of the patients; clinical features were similar in the groups with and without these variants, suggesting that unexplored regions of the ABCB4 gene or different genes could be involved [6].
The ABCB11 gene encodes the bile salt export pump (BSEP), which transports bile salts across the canalicular membrane of the hepatocyte, the limiting step in bile secretion [16]. Mutations in BSEP are linked to PFIC2, BRIC2, ICP, and drug-induced liver injury (DILI) [5, 17, 18]. Two BSEP mutations have also been identified in patients with primary intrahepatic stones [19], although not corresponding to LPAC syndrome. Of note, in contrast to most MDR3-related diseases (except for most cases of BRIC3 [8]), BSEP-related diseases are characterized by normal GGT activity, due to low levels of bile salts in bile and consequently less damage do cholangiocytes [18].
We present a case of persistent jaundice in the setting of both ABCB4 and ABCB11 mutations, whose interpretation is not straightforward. There is a clear contribution of gallstone disease, fulfilling the criteria for LPAC: age of presentation below 40 years and hepatolithiasis; this diagnosis is further supported by a family history of gallstone disease in a first-degree relative and by a favorable response to UDCA.
The persistent jaundice despite retrieval of biliary stones raises the possibility that a second mechanism might have been involved, namely DILI. Indeed, the abrupt increase in total bilirubin occurred 4 days after the introduction of ciprofloxacin, which has been described as a cause of cholestatic hepatitis with high bilirubin [20]. Since the rise in bilirubin happened 24 h after the first ERCP, propofol could also be implicated. However, DILI is not frequent with such short anesthesia and it is more often hepatocellular; also, there was no effect on re-exposure (third ERCP, Fig. 3). To our knowledge, iobitridol has no known liver toxicity. Two hypotheses may be considered: either there was indeed superimposed DILI (to which our patient has increased susceptibility, as explained below), most likely related to ciprofloxacin, or there was just a prolonged cholestatic response to the initial insult (which would be cholangitis due to choledocholithiasis), with bilirubin peaking after the other liver enzymes as happens in other types of liver injury. In the latter case, this prolonged response could be explained in the setting of the gene defects discussed below.
The ABCB4 mutation found in our patient (homozygous Arg652Gly) is a missense mutation that, although probably representing a polymorphism [12], has been linked to ICP [11] and PFIC3 [8]. It is thought to have mild consequences under normal conditions but possibly becomes clinically relevant during pregnancy or when combined with another mutation [8, 12, 13] (which is the case in our patient); however, it has not been linked to LPAC. The ABCB11 mutation (heterozygous Val444Ala) is a common variation (allele frequencies 50–93%) [21], known to cause a 3-fold increase in the risk of DILI [13]; it was also implicated in ICP and contraceptive-induced cholestasis [22]. This missense mutation probably induces protein misfolding, increasing its degradation [18]. To our knowledge, this has also not been associated with LPAC.
The mechanisms involved in the interplay between these two mutations and their contribution to our patients evolution are uncertain. Other cases of complex interaction between ABCB4 and ABCB11 mutations have been described (specifically Val444Ala mutation, as in our patient): in a patient with ICP, the combination of ABCB4 and homozygous ABCB11 Val444Ala mutations was though to explain the severity and early onset of the disease; it was speculated that the reduction in MDR3 expression would modify the phospholipid composition of the hepatocyte canalicular membrane, thereby altering BSEP expression [23]. In a pair of siblings with MDR3 mutations and complex cholestatic disorders, the heterozygous Val444Ala mutation was considered to be a disease modifier [24]. Recently, this ABCB11 mutation was also thought to induce decompensation in a patient with a mild form of PFIC2 exposed to azithromycin [25].
In summary, we describe the unique case of an adult male with acute-onset jaundice (high GGT), with choledocholithiasis, hepatolithiasis, and persistent conjugated hyperbilirubinemia after retrieval of stones, fulfilling the criteria for LPAC syndrome and with possible superimposed DILI, in whom ABCB4 and ABCB11 mutations were found, both of which had not been previously described in association with LPAC. This case illustrates the complexity of the genetics of cholestatic diseases, a field of intense research in the last years, where many questions remain unsolved.
References
1 Rosmorduc O, Hermelin B, Poupon R: MDR3 gene defect in adults with symptomatic intrahepatic and gallbladder cholesterol cholelithiasis. Gastroenterology 2001;120:1459–1467. [ Links ]
2 Rosmorduc O, Poupon R: Low phospholipid associated cholelithiasis: association with mutation in the MDR3/ABCB4 gene. Orphanet J Rare Dis 2007;2:29. [ Links ]
3 Rosmorduc O, Hermelin B, Boelle PY, Parc R, Taboury J, Poupon R: ABCB4 gene mutation-associated cholelithiasis in adults. Gastroenterology 2003;125:452–459. [ Links ]
4 Erlinger S: Low phospholipid-associated cholestasis and cholelithiasis. Clin Res Hepatol Gastroenterol 2012;36(suppl 1):S36-S40. [ Links ]
5 Nicolaou M, Andress EJ, Zolnerciks JK, Dixon PH, Williamson C, Linton KJ: Canalicular ABC transporters and liver disease. J Pathol 2012;226:300–315. [ Links ]
6 Poupon R, Rosmorduc O, Boëlle PY, et al: Genotype-phenotype relationships in the low-phospholipid-associated cholelithiasis syndrome: a study of 156 consecutive patients. Hepatology 2013;58:1105–1110. [ Links ]
7 Ruetz S, Gros P: Phosphatidylcholine translocase: a physiological role for the mdr2 gene. Cell 1994;77:1071–1081. [ Links ]
8 Davit-Spraul A, Gonzales E, Baussan C, Jacquemin E: The spectrum of liver diseases related to ABCB4 gene mutations: pathophysiology and clinical aspects. Semin Liver Dis 2010;30:134–146. [ Links ]
9 Deleuze JF, Jacquemin E, Dubuisson C, et al: Defect of multidrug-resistance 3 gene expression in a subtype of progressive familial intrahepatic cholestasis. Hepatology 1996;23:904–908. [ Links ]
10 Brenard R, Geubel AP, Benhamou JP: Benign recurrent intrahepatic cholestasis. A report of 26 cases. J Clin Gastroenterol 1989;11:546–551. [ Links ]
11 Floreani A, Carderi I, Paternoster D, et al: Intrahepatic cholestasis of pregnancy: three novel MDR3 gene mutations. Aliment Pharmacol Ther 2006;23:1649–1653. [ Links ]
12 Jacquemin E, De Vree JMML, Cresteil D, et al: The wide spectrum of multidrug resistance 3 deficiency: from neonatal cholestasis to cirrhosis of adulthood. Gastroenterology 2001;120:1448–1458. [ Links ]
13 Lang C, Meier Y, Stieger B, et al: Mutations and polymorphisms in the bile salt export pump and the multidrug resistance protein 3 associated with drug-induced liver injury. Pharmacogenet Genomics 2007;17:47–60. [ Links ]
14 Ziol M, Barbu V, Rosmorduc O, et al: ABCB4 Heterozygous gene mutations associated with fibrosing cholestatic liver disease in adults. Gastroenterology 2008;135:131–141. [ Links ]
15 Smith AJ, Van Helvoort A, Van Meer G, et al: MDR3 P-glycoprotein, a phosphatidylcholine translocase, transports several cytotoxic drugs and directly interacts with drags as judged by interference with nucleotide trapping. J Biol Chem 2000;275:23530–23539. [ Links ]
16 Stieger B, Beuers U: The canalicular bile salt export pump BSEP (ABCB11) as a potential therapeutic target. Curr Drug Targets 2011;12:661–670. [ Links ]
17 Lang T, Haberl M, Jung D, et al: Genetic variability, haplotype structures, and ethnic diversity of hepatic transporters MDR3 (ABCB4) and bile salt export pump (ABCB11). Drug Metab Dispos 2006;34:1582–1599. [ Links ]
18 Kubitz R, Dröge C, Stindt J, Weissenberger K: The bile salt export pump (BSEP) in health and disease. Clin Res Hepatol Gastroenterol 2012;36:536–553. [ Links ]
19 Pan S, Li X, Jiang P, et al: Variations of ABCB4 and ABCB11 genes are associated with primary intrahepatic stones. Mol Med Rep 2015;11:434–446. [ Links ]
20 Sherman O, Beizer JL: Possible ciprofloxacin-induced acute cholestatic jaundice. Ann Pharmacother 1994;28:1162–1164. [ Links ]
21 Ho RH, Leake BF, Kilkenny DM, et al: Polymorphic variants in the human bile salt export pump (BSEP; ABCB11): functional characterization and interindividual variability. Pharmacogenet Genomics 2010;20:45–57. [ Links ]
22 Meier Y, Zodan T, Lang C, et al: Increased susceptibility for intrahepatic cholestasis of pregnancy and contraceptive-induced cholestasis in carriers of the 1331T>C polymorphism in the bile salt export pump. World J Gastroenterol 2008;14:38–45. [ Links ]
23 Keitel V, Vogt C, Häussinger D, Kubitz R: Combined mutations of canalicular transporter proteins cause severe intrahepatic cholestasis of pregnancy. Gastroenterology 2006;131:624–629. [ Links ]
24 Poupon R, Barbu V, Chamouard P, Wendum D, Rosmorduc O, Housset C: Combined features of low phospholipid-associated cholelithiasis and progressive familial intrahepatic cholestasis 3. Liver Int 2010;30:327–331. [ Links ]
25 Waisbourd-Zinman O, Surrey LF, Schwartz AE, Russo PA, Wen J: A rare BSEP mutation associated with a mild form of progressive familial intrahepatic cholestasis type 2. Ann Hepatol 2017;16:465–468. [ Links ]
Statement of Ethics
This study did not require informed consent nor review/approval by the appropriate ethics committee.
Disclosure Statement
The authors have no conflicts of interest to declare.
* Corresponding author.
Dr. Mariana Ferreira Cardoso
Gastroenterology Department
Hospital Professor Doutor Fernando Fonseca, IC-19, Venteira
PT–2720-276 Amadora (Portugal)
E-Mail marianafcardoso@gmail.com
Received: September 7, 2017; Accepted after revision: October 25, 2017
Author Contributions
Drafting of the article: Mariana Ferreira Cardoso. Critical revision of the article: Joana Carvalho e Branco, Rita Carvalho. Final approval of the version to be published: Mariana Ferreira Cardoso, Joana Carvalho e Branco, Vera Anapaz, Catarina Graça Rodrigues, Rita Carvalho, David Horta, Alexandra Martins, Jorge Reis.

