











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroduction
Genetic testing aims to find a variant known to be clinically actionable, meaning the management of the patient will be altered according to the presence of a variant [1]. It may include a single-gene analysis or a comprehensive multi-gene cancer panel. Based on next-generation sequencing technology, the latter allows for the simultaneous analysis of many genes associated with multiple cancers or syndromes [1].
In the last decade, there was a significant increase in the request of broader panels of genes as multi-gene panel testing became widely available and commonly used in the study of hereditary cancer syndromes, particularly in breast, ovarian and endometrial cancers [2]. Knowledge of genetic susceptibility to gastrointestinal cancers has also increased thanks to evolving research, identification of new gene variants and clarification of the role of previously known variants [3].
Patients with hereditary gastrointestinal cancer syndromes are at high risk of developing one or more malignancies, synchronously or metachronously, in several organs (stomach, small bowel, colon, rectum, pancreas, endometrium, ovaries, breast,...) [3]. It is therefore of paramount importance to identify patients and family members carrying a genetic predisposition for gastrointestinal cancer, so they can be offered screening (to prevent or detect cancer at an earlier stage) and risk-reducing measures (counselling about lifestyle habits, chemoprevention or prophylactic surgeries) [4].
The present article aims to: (1) discuss the advantages and the drawbacks related to multi-gene panel testing, particularly in the setting of Gastrointestinal Familial Cancer Risk clinic; and (2) address the available recommendations on management of individuals with increased risk of gastrointestinal cancer based on germline variant status.
Methods
A narrative non-systematic review was performed based on an electronic search through the medical literature using PubMed and Google. The keywords “Multi-gene panel testing,” “Next-generation sequencing,” “Hereditary colorectal cancer,” “Hereditary pancreatic cancer,” “Hereditary gastric cancer,” “Hereditary gastrointestinal cancer,” “Polyposis syndromes” were used. Review articles and guidelines from gastroenterology, endoscopy, oncology and genetics were included. When more than one guideline concerning the same subject was available, the most up-to-date one was selected. Articles published in English were considered.
Multi-Gene Panel Testing
In recent years, there was a massive spread in the use of multi-gene panels in order to identify patients at risk for hereditary cancer.
Single-gene analysis may be suitable when a specific disease is known to be associated with only one gene or when testing an individual from a family with a known pathogenic variant. However, when more than one gene may explain the patient’s clinical presentation and family history, selecting a multi-gene panel testing may be more useful as it provides more information in a sole analysis [5, 6]. A panel testing allows not only to identify expected pathogenic alterations in gene(s) known to cause the phenotype for which the sequencing was ordered, but also to search for pathogenic or likely pathogenic variants that may or may not be related with the indication of the test [7].
Furthermore, multi-gene panels are cost-effective and time efficient. Compared to several sequential single gene testings, a multi-gene panel becomes more affordable and prevents patient’s fatigue and potentially loss of follow-up from multiple testings [6, 8].
The National Comprehensive Cancer Network (NCCN) guideline [5] proposes a set of clinical scenarios for which multi-gene panel testing should be considered, namely:
when personal medical and/or family cancer history meets criteria for more than one hereditary cancer syndrome
when family cancer history does not meet established testing guidelines, but consideration of inherited cancer risk persists
in individuals concerned about cancer predisposition for whom family cancer history is limited or unknown
A multi-gene panel is extensively used for the study of hereditary breast and ovarian cancer. It includes genes like ATM, BRCA1, BRCA2, CHEK2, MLH1, MSH2, MSH6, PALB2, PTEN, STK11or TP53 [9]. These genes also share a susceptibility to gastrointestinal cancer, requiring a specific management and follow-up [5, 10].
In the Gastroenterology setting, a multi-gene panel testing approach may also be helpful when alterations in several genes (frequent or infrequent) may explain a patient or family phenotype. For example, hereditary colorectal cancer and/or polyposis may be caused by pathogenic variants in many genes (APC, MUTYH, SMAD4, BMPR1A, STK11, PTEN, MLH1, MSH2, MSH6, PMS2, EPCAM, TP53, AXIN2, GREM1, MSH3, NTHL1, POLE, POLD1 or GALNT12) [5, 9]. Likewise, the study of hereditary pancreatic cancer may require the analysis of several genes (CDKN2A, STK11, ATM, BRCA1, BRCA2, PALB2, MLH1 or MSH2) [10].
On the other hand, multi-gene panels present some limitations. They increase the likelihood of identifying pathogenic variants for which clinical management is uncertain due to lack of data regarding degree of cancer risk [5, 8]. They also increase the risk of finding variants of uncertain significance that are not actionable [5, 8]. Consequently, genetic counselling becomes more complex, since physicians may not know how to communicate and manage risk for carriers of these gene variants, beyond family history. Ultimately, multi-gene panel testing may cause overtreatment or overscreening, as well as undue anxiety and cancer worry, with negative implications in the patient’s quality of life. It may also result in a work overload and increasing waiting list for endoscopy and radiology units. However, these challenges may be minimized by restricting the variants to be reported to those that are known or expected to cause the disorder [7].
Panel Gene Testing in Gastroenterology
In our clinical practice, several patients have been referred to our Gastrointestinal Familial Cancer Risk clinic for management of gene variants associated with a potentially increased risk of digestive cancer. Many of these variants were detected in multi-gene panel testing primarily requested for study of hereditary breast, ovarian or endometrial cancer, including patients with personal and/or family history of cancer.
Examples of genes currently included on our sequencing multi-gene panel are: ATM, AXIN2, BRCA1, BRCA2, BMPR1A, CDH1, CDKN2A, CHEK2, EPCAM, GALNT12, GREM1, MLH1, MSH2, MSH3, MSH6, MUTYH, NTHL1, PMS2, PALB2, POLE, POLD1, PTEN, SMAD4, STK11or TP53 [2, 6, 8]. There has been some progress in understanding the clinical relevance of variants in many of these genes, allowing the establishment of consensus recommendations to guide management of carriers [5, 10].
Pathogenic variants in several genes are known for causing specific hereditary disorders or syndromes associated with increased risk of digestive cancer. The most prevalent affected genes and syndromes are the following: monoallelic pathogenic variants in a DNA mismatch repair gene (MLH1, MSH2, MSH6, PMS2) or deletions of the 3′ end of the EPCAM gene are responsible for Lynch syndrome [5], while biallelic variants lead to Constitutional Mismatch Repair Deficiency syndrome [5]; APC and MUTYH pathogenic variants are responsible for Familial adenomatous polyposis and MUTYH-associated polyposis (biallelic), respectively [5]; SMAD4 and BMPR1A pathogenic variants are identified in patients with Juvenile polyposis syndrome [5]; pathogenic variants in STK11 and PTEN are associated with Peutz-Jeghers syndrome and Cowden syndrome, respectively [5]; TP53 pathogenic variants are responsible for Li-Fraumeni syndrome [5]; CDH1 and CTNNA1 pathogenic variants are associated with Hereditary diffuse gastric cancer [11]; CDKN2A pathogenic variants characterize the Familial atypical multiple mole and melanoma syndrome, associated with an increased risk of pancreatic cancer [10].
Specific guidelines concerning gastrointestinal cancer screening are widely available for these established conditions [5, 12-14].
Other less prevalent germline variants associated with a potentially increased risk for one or more gastrointestinal cancer types have been described:
The CHEK2 (checkpoint kinase 2) gene is a tumor suppressor gene, located on chromosome 22, and it is involved in DNA repair [2]. Pathogenic variants in CHEK2 are associated with a moderate risk for breast and colorectal cancer [5, 12].
BRCA1 and BRCA2 are tumor suppressor genes, located on chromosomes 17 and 13 (respectively), and participate in homologous recombination repairing of damaged DNA [8]. They are the most frequent genes involved in hereditary breast and ovarian cancer and they also increase the risk for pancreatic cancer [12]. Pathogenic variants in BRCA2 are also associated with an increased risk of gastric cancer [11].
PALB2 (partner and localizer of BRCA2) is a tumor suppressor gene located on chromosome 16 [8]. Pathogenic variants in PALB2 are associated with breast, gastric and pancreatic cancer [11, 12].
ATM (ataxia-telangiectasia mutated) gene is located on chromosome 11 and plays a role in cell division and DNA repair [8]. Pathogenic variants in ATM are associated with increased risk for breast, gastric and pancreatic cancer [11, 12].
GREM1 (Gremlin 1) is a gene located on chromosome 15 and it is a BMP antagonist [15]. Pathogenic variants are associated with colorectal polyposis and cancer [5].
POLE (DNA Polymerase Epsilon, Catalytic Subunit) and POLD1 (DNA Polymerase Delta 1, Catalytic Subunit) are genes involved in DNA replication and repair, and they are located on chromosomes 12 and 19, respectively. Pathogenic variants in POLE and POLD1 may be associated with predisposition to colorectal polyposis and cancer [5].
GALNT12 (polypeptide N-acetylgalactosaminyltransferase 12) is located on chromosome 9 and may play a role in glycosylation of mucin-type glycans [16]. Pathogenic variants in this gene are associated with an increased susceptibility to colorectal cancer [5].
AXIN2 is a gene involved in the regulation of the stability of beta-catenin in the Wnt signaling pathway and is located on the chromosome 17 [17]. Pathogenic variants in AXIN2 are associated with colorectal cancer [5].
The NTHL1 gene is located on the chromosome 16 and is involved in the DNA base excision repair pathway [18]. Biallellic pathogenic variants in this gene are associated with an increased risk for colorectal polyposis and cancer [5].
MSH3 is a DNA mismatch repair gene located on the chromosome 5, and biallelic pathogenic variants also present predisposition for colorectal polyposis and cancer [5].
Particularly in the setting of colorectal cancer and/or polyposis, the Collaborative Group of the Americas on Inherited Gastrointestinal Cancer proposes the use of multi-gene panel testing in all patients with colorectal cancer diagnosed under the age of 50 years, in patients with colorectal cancer and at least one first degree relative with colorectal or endometrial cancer, and in patients with at least 10 cumulative adenomatous polyps or three cumulative hamartomatous polyps [19].
Management of New Germline Variants in Gastroenterology
Identification of patients and families with genetic predisposition for digestive cancer is of great importance as endoscopic and/or radiological surveillance can help prevent the development of cancer or its detection at an earlier stage [14]. However, evidence regarding the clinical impact of these less prevalent gene variants on the development of gastrointestinal malignancies are still limited, whereby recommendations about management are also scarce. In Table 1, we summarize the available guidelines on management of individuals with increased risk of gastrointestinal cancer based on germline variant status.
Table 1: Screening recommendations on management of individuals with increased risk of gastrointestinal cancer based on germline variant status
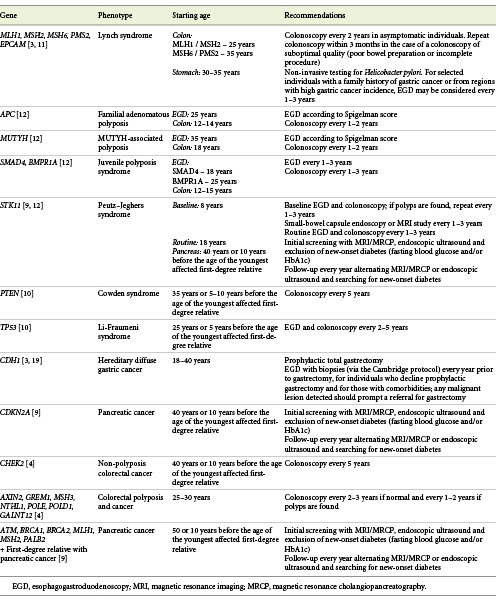
As genetic testing and multi-gene panels are rapidly evolving with an increasingly number of germline pathogenic variants detected, it is of paramount importance that patients receive adequate post-test counselling and follow-up. Gastroenterologists involved in the care of patients with gastrointestinal cancer should be familiar with hereditary cancer syndromes and main germline variants associated with cancer susceptibility. Referral for expert centers should be warranted for patients or their relatives (carriers) in order to provide adequate genetic counselling and surveillance [3].
Below, we describe the available recommendations on management of these new less prevalent germline pathogenic variants.
Colorectal Cancer
CHEK2 pathogenic variants are associated with non-polyposis colorectal cancer and, for their carriers, NCCN recommends colonoscopy every 5 years beginning at age 40 years or, if there is history of a first-degree relative with colorectal cancer, starting 10 years before the age of the youngest affected blood relative [5]. These recommendations are identical to the guidelines regarding screening in patients with a family history of colorectal cancer.
For carriers of pathogenic variants in AXIN2, GREM1, MSH3, NTHL1, POLE, POLD1 and GALNT12 genes, associated with colorectal adenomatous polyposis and cancer, NCCN recommends starting colonoscopy screening at the age of 25-30 years, with follow-up colonoscopies every 2-3 years if negative, and 1-2 years if polyps are found [5]. Surgery may be considered if the polyp burden becomes unmanageable by colonoscopy [5].
Pancreatic Cancer
Pathogenic variants in ATM, BRCA1, BRCA2, MLH1, MSH2 and PALB2 present an increased susceptibility for pancreatic cancer, and the International Cancer of the Pancreas Screening (CAPS) Consortium recommends pancreas cancer screening for those carriers who also have a first-degree relative with pancreatic cancer [10]. Screening should begin at the age of 50 years or 10 years before the age of the youngest affected blood relative with magnetic resonance cholangiopancreatography (MRCP), endoscopic ultrasound and exclusion of new-onset diabetes (fasting blood glucose and/or HbA1c) [10]. Thereafter, the CAPS Consortium recommends yearly follow-up alternating MRCP or endoscopic ultrasound and searching for new-onset diabetes [10]. Discontinuation of pancreas cancer screening should be considered if a patient becomes unfit for a potential surgical resection [20].
Conclusion
Multi-gene panel testings have been increasingly used to study potentially hereditary cancer phenotypes, leading to detection of less prevalent pathogenic gene variants associated with predisposition to gastrointestinal cancer. However, evidence concerning penetrance and clinical impact of these genetic alterations are still limited, hampering the development of recommendations on patients and carriers’ management.
In this review, we approached the impact of multi-gene panel testing in gastroenterology and present the available recommendations on the management of individuals with increased risk of gastrointestinal cancer based on germline variant status.
As knowledge of genetic susceptibility to gastrointestinal cancers expands and more variants are detected thanks to multi-gene panels, the results pose challenges for both physicians and carriers (patients or family members). For physicians, interpretation of the results becomes more complex as it requires understanding of the genetic susceptibility to cancer risk (particularly the penetrance of the new less prevalent gene variants) and careful evaluation of the carrier (including personal and family history) in order to propose a management plan. Variants carriers seek appropriate counselling to balance the benefits and the psychological burden of cancer screening and risk-reducing procedures.

