











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
Introduction
Sickle cell disease (SCD) is a common hemoglobinopathy that can affect multiple organ systems, including the gastrointestinal tract [1]. The hepatobiliary system is one of the most common intra-abdominal organs involved in SCD, with hepatic involvement in 10-40% of sickle cell crises. Manifestations range from mild hyperbilirrubinemia to acute liver failure, and it is a rare condition that represents one of the most challenging of all critical pediatric illnesses [2].
Case Presentation
A 7-year-old African boy with a history of homozygous HbS SCD presented to the emergency department with a 24-h history of fever, increasing abdominal pain and vomiting, chest pain, and dry cough. His SCD was previously complicated by recurrent pain crises requiring hospitalization and cholelithiasis, although there were no clinical, biochemical, or imaging features of chronic liver disease prior to this event. His medication included hydroxyurea 500 mg and folic acid 5 mg daily. He had no other comorbidities.
On physical examination, the patient was drowsy (grade I hepatic encephalopathy according to West Haven criteria), ill-appearing, pale, and dehydrated. He was afebrile, with an adequate blood pressure of 114/55 mm Hg, a pulse rate of 120 beats/min, a capillary refill time of 4 s, a respiratory rate of 30 breaths/min, and oxygen saturation of 94% on room air. Breath sounds were clear to auscultation bilaterally. Heart sounds were normal with no audible murmurs and a regular rate and rhythm. There was right-upper-quadrant tenderness and the liver edge was palpable 2 cm below the right costal margin. The spleen was nonpalpable. The remaining systemic examination was normal.
Initial laboratory evaluation demonstrated hemoglobin 5.6 g/dL (baseline hemoglobin 8-8,5 g/dL), leukocytes 9,500/µL-neutrophils 7,200/µL (75.3%), platelets 131,000/µL, serum glucose 27 mg/dL, creatinine 1.0 mg/dL, blood urea 49.5 mg/dL, sodium 138 mmol/L, potassium 4.9 mmol/L, aspartate aminotransferase (AST) 200 IU/L, alanine aminotransferase (ALT) 48 IU/L, total bilirubin 11.8 mg/dL, direct bilirubin 2.8 mg/dL, lactate dehydrogenase (LDH) 1,249 IU/L, and C-reactive protein 18.95 mg/dL. Blood gas analysis showed metabolic acidosis, with a venous pH of 7.01, venous partial pressure of carbon dioxide 32.1 mm Hg, venous bicarbonate 8.1 mmol/L, base excess -22.7 mmol/L, and venous lactate 16 mmol/L. There was no pulmonary opacity on the chest X-ray.
Initially, he received fluid resuscitation with normal saline bolus (10 mL/kg) and then red blood cell transfusion (5 mL/kg). The hypoglycemia was corrected with dextrose 10% bolus and ceftriaxone was empirically started. However, his clinical picture progressively worsened, with indicators of respiratory distress like tachypnea, retractions of the muscles of the chest wall, and grunting, so he was transferred to the Pediatric Intensive Care Unit (PICU) for further management.
On the first day of admission in PICU, he became progressively more lethargic and had increasing abdominal pain and jaundice but remained hemodynamically stable (MAP >75 mm Hg) with spontaneous breathing, without a need for organ support. His blood analysis showed a significant worsening: AST 9,472 IU/L, ALT 2,683 IU/L, lactate dehydrogenase 10,621 IU/L, total bilirubin 15.4 mg/dL (from which >50% was conjugated bilirubin - 8.69 mg/dL), associated with coagulopathy (international normalized ratio [INR] 5.16 and partial thromboplastin time 94.8/29 s) not corrected by vitamin K administration (INR 3.26), and persistent hypoglycemia despite a high glucose infusion rate (maximum 10 g/kg/day). Ammonia ranged from 71 to 63 µmol/L (N: <60). Other values showing diminished hepatic synthesis included low factor V (10%, N: 50-100), factor VII (28%, N: 70-130), fibrinogen (67 mg/dL) and albumin (2.6 g/dL), with normal factor VIII (94%, N: 50-150). He continued to have kidney injury with a creatinine of 0.68 mg/dL (baseline creatinine 0.22-0.31 mg/dL), and blood urea 57 mg/dL. Peripheral blood smear showed anisopoikilocytosis, hypochromia, multiple sickle cells, and Howell-Jolly bodies.
The etiological investigation was negative and included toxicology screening (in the urine and paracetamol serum level); serologic screening for viral hepatitis (i.e., HAV, HBV, HCV, and HEV), Epstein-Barr virus, Cytomegalovirus, Coxsackievirus, Echovirus, Parvovirus, and Herpes simplex virus types 1 and 2; antinuclear-, smooth-muscle, and liver-kidney-microsomal antibodies; ceruloplasmin; and alpha-1 antitrypsin. Abdominal ultrasound revealed hepatomegaly and cholelithiasis, but there was no evidence of intra- or extrahepatic biliary duct obstruction or dilation and there were no signs of chronic liver disease. The liver vascular ultrasound showed patent hepatic vasculature and appropriate blood flow. Ceftriaxone was changed to cefotaxime due to cholestasis.
The case was discussed by the multidisciplinary team (PICU, Hematology, and Hepatology), the diagnosis of SCIC was considered, and exchange transfusion was decided. For this purpose, a central catheter was placed in the patient’s right jugular vein. Exchange transfusion was performed using repeated alternating isovolumetric phlebotomy and blood transfusion, with a subsequent HbS of 13.2%.
Over the ensuing days, there was a quick favorable evolution of his clinical and laboratory status. The patient was discharged from the PICU to the Hematology ward 4 days after the exchange transfusion. His blood analysis showed a significant improvement: hemoglobin 9.5 g/dL, AST 178 IU/L, ALT 377 IU/L, total bilirubin 7.24 mg/dL, INR 1.33, partial thromboplastin time 37.8/29 s, creatinine 0.39 mg/dL, and blood urea 25 mg/dL. Blood culture was positive for Streptococcus pneumoniae and cefotaxime was changed to ampicillin. The rest of his course was unremarkable.
Discussion
Sickle cell hepatopathy is an inclusive term encompassing the different kinds of liver disease found in patients with SCD [3]. The incidence of sickle hepatopathy is difficult to establish because abnormalities in liver function tests are common in these patients [4]. A spectrum of clinical manifestations may be observed for the same underlying pathophysiology, i.e., sinusoidal obstruction by sickled red blood cells resulting in hepatocyte ischemia, with secondary ballooning of adjacent hepatocytes and intracanalicular cholestasis. Depending on the relative degrees of ischemia, cholestasis, and cell trapping, the crisis may present as an acute sickle cell hepatic crisis, hepatic sequestration, or SCIC [5]. Classification can be difficult due to the overlapping diagnostic criteria (Table 1) [2, 6, 7].
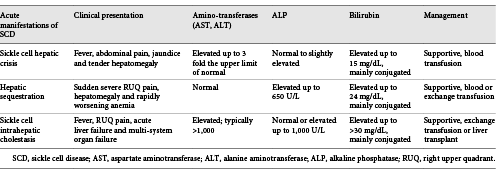
SCIC is one of the rarest and the most severe acute hepatic manifestations of SCD and a mortality rate of approximately 40% [6]. It presents initially as a severe acute hepatic crisis with fever, leukocytosis, right-upper-quadrant abdominal pain, and jaundice, but can rapidly progress to multiple organ dysfunction including acute liver failure and acute kidney injury, as was seen in our patient [8]. Biochemical evidence showed significantly elevated bilirubin levels, which were mainly due to a rise in conjugated component; acute liver injury with transaminase levels >1,000 mg/dL, reflecting severe ischemic injury; and derangement of coagulation profile in the form of elevated prothrombin time, INR, and partial thromboplastin time [2, 6, 7]. SCIC is most commonly described in its acute form, but it can present as recurrent episodes, become chronic, and eventually evolve to progressive liver failure. To date, only 7 cases of chronic SCIC have been described in adults, and this entity is not well characterized [9]. To the best of our knowledge, there is no data on the incidence of recurrent or chronic SCIC in the pediatric setting.
An extensive workup for acute liver injury was undertaken and competing etiologies were excluded (viral infectious diseases, autoimmune diseases, Wilson disease, and alpha-1 antitrypsin deficiency). Sepsis-associated liver dysfunction (with later identification of S. pneumoniae) was also considered. During sepsis, not only infection itself, but also hyperactivity of the inflammatory response and microcirculatory failure are responsible for liver injury. Sepsis is almost invariably associated with coagulation abnormalities ranging from subclinical activation of blood coagulation to acute disseminated intravascular coagulation. The main cause of coagulopathy in sepsis is microvascular endothelial injury, resulting in an imbalance between fibrinolysis and coagulation, that leads to the consumption and subsequent depletion of platelets and coagulation factors (including factor VIII, which was normal in our patient). Also, in pediatric septic shock, defined as severe infection leading to cardiovascular dysfunction (including hypotension, the need for treatment with a vasoactive medication, or impaired perfusion) [10], hypoxemia/liver hypoperfusion could explain hypoxic hepatitis and hyperbilirubinemia, as energy shortage during this process would impair most bile synthesis steps [11]. However, our patient was always hemodynamically stable, and there were no signs of decreased hepatic blood flow that would support this hypothesis. Finally, there was a significant improvement after the exchange transfusion treatment that reversed the acute liver injury, which would support the diagnosis of SCIC. Complications due to biliary disease such as cholecystitis and choledocholithiasis were excluded on abdominal ultrasound [12]. Although liver biopsy is virtually diagnostic of SCIC, it has been associated with serious complications when performed in patients with acute sickle cell hepatopathy, so the risk-benefit ratio must be considered. In our case, it was not performed in the setting of severe coagulopathy [13, 14].
As in this case, SCIC can rapidly progress to acute liver failure, defined as biochemical evidence of liver injury in a child without evidence of chronic liver disease, coagulopathy not corrected by vitamin K administration, and INR >1.5 if the patient has encephalopathy or >2.0 if encephalopathy is absent. This a life-threatening condition due to the unpredictable and potentially devastating complications as well as multiple organ system involvement. Proper management is dependent on intensive collaborative clinical care and implementing a timely and appropriate therapy [15, 16].
In patients with SCIC, therapy is aimed at aggressive supportive measures, including exchange transfusion to maintain a low level of HbS (<30%) [17, 18]. If supportive measures fail, patients are likely to have a worse prognosis, and liver transplantation remains the only viable therapeutic option. However, experience of liver transplantation for hepatic complications of SCD is limited; only 23 cases have been reported in the literature, with a survival rate of 66% in pediatric patients [6] and an increased risk of a vaso-occlusive crisis [19, 20]. In this patient, we performed a manual exchange transfusion, which reversed the acute liver failure and avoided the need for a liver transplant. This is a time-consuming procedure, that needs skilled staff and constant medical supervision, and should only be performed in specialized units. Temporary renal replacement may be required for acute kidney injury, whose causes include acute tubular necrosis, hypovolemia, sepsis, nephrotoxic medications, and functional renal failure. We believe that, in this case, the acute kidney injury was the result of circulatory changes secondary to liver failure as it rapidly improved with the correction of the hepatic dysfunction [21, 22].
In summary, when a patient with SCD presents with abdominal pain and extreme hyperbilirubinemia, a careful and prompt evaluation is necessary to establish a diagnosis of sickle cell hepatopathy. Physical examination, laboratory workup, and appropriate diagnostic imaging are needed to diagnose and characterize sickle cell hepatopathy and exclude other conditions. This case shows one of the rarest and most extreme acute hepatic manifestations of SCD and reinforces that exchange transfusion is an effective treatment for SCIC. With the exchange transfusion, we were able to reverse the acute liver failure and change the outcome of a potentially fatal condition.

