











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
Introduction
Pancreatic neuroendocrine tumours (PNETs) are neoplasms that arise from the islet cells of the pancreas, and these tumours account for 1-3 % of pancreatic neoplasms. PNETs are categorised as functioning or non-functioning tumours based on their ability to secrete specific hormones resulting in clinical symptoms [1]. The non-functional PNETs are commonly detected incidentally on imaging or from symptoms due to the mass effect of the tumour. Insulinoma, glucagonoma, gastrinoma, somatostatinoma are the few common examples of functioning PNETs with variable presentation depending on the overproduced hormones [2]. However, ectopic secretion of adrenocortico-tropic hormone (ACTH) from PNET causing Cushing syndrome is rare, with only a few cases reported worldwide [1].
Case Report/Case Presentation
A 29-year-old lady presented with excessive weight gain, proximal muscle weakness, facial acne, and amenorrhea for 6 months. She has no family history of endocrine-related malignancy. Clinical examination revealed cushingoid appearance, central obesity, purplish striae at the abdomen and both arms. Her blood pressure was 155/99 mm Hg, and her body mass index was 54. She was diagnosed of having hypertension and diabetes mellitus on her admission. 24-hour urinary cortisol was 1,649.9 nmol/L and serum cortisol was 656.8 nmol/L on an overnight dexamethasone suppression test. A diagnosis of Cushing syndrome was made after magnetic resonance imaging of the brain excluded pituitary disease. Computed tomography of the adrenal found an illdefined hypodense lesion at the body of pancreas measuring 3.7 × 5.5 cm (Fig. 1a) with atrophic pancreatic tail and dilated distal pancreatic duct (Fig. 1b). In addition, there were ill-defined hypodense lesions in the liver suggestive of metastases (Fig. 1c) with normal adrenal glands bilaterally. Gallium-68 dotatate scan revealed heterogeneous uptake at the pancreatic body lesion suggesting somatostatin receptor avid primary disease. Endoscopic ultrasonography (EUS) showed an ill-defined hypoechoic lesion at the body of pancreas (Fig. 1d). Fine-needle aspiration cytology performed on the lesion yielded well-differentiated tumour cells on cell block (Fig. 2a, H&E staining, ×400). Immunohistochemistry study showed these cells were positive for chromogranin (Fig. 2b) and synaptophysin (Fig. 2c), CKAE1/AE3 (Fig. 2d) as well as ACTH (Fig. 2e, ×200) confirming ACTH-secreting pancreatic neuroendocrine tumour. Ki-67 proliferative index was 5% with no mitotic figure seen (Fig. 2f), indicating PanNET G2 on World Health Organisation (WHO) 2017 classification and grading of pancreatic neuroendocrine neoplasm with T2N0M1 based on TNM staging. She was commenced on oral ketoconazole to suppress her hypercortisolism. A multidisciplinary meeting had agreed on the consensus that distal pancreatectomy would be the most suitable option to control her disease. Unfortunately, she refused surgical resection and defaulted her follow-up and medications before succumbing to sepsis a few months later.
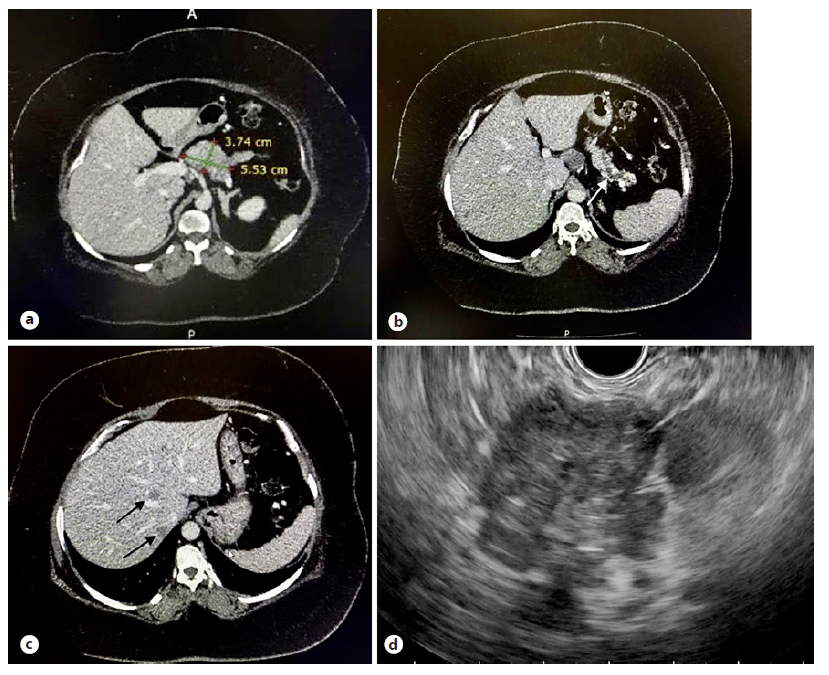
Fig. 1 CT showed an ill-defined hypodense lesion at the body of pancreas (a) causing main pancreatic duct dilatation with pancreatic tail atrophy (white arrow, b) and small hypodense liver lesions suggestive of metastases (black arrows, c). An ill-defined hypoechoic lesion at the body of pancreas was visualised on EUS (d).
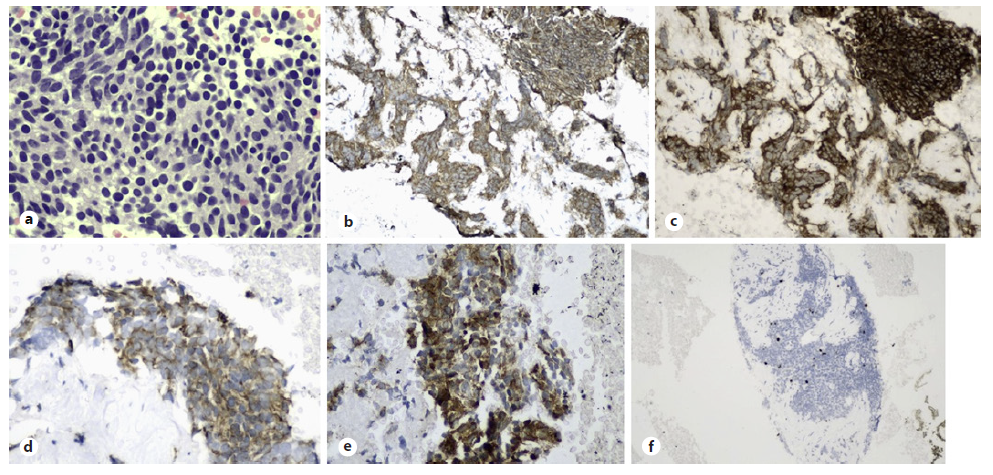
Fig. 2 Well-differentiated tumour cells were seen on cell block (a, H&E staining, ×400). These cells were positive for chromogranin (b) and synaptophysin (c), CKAE1/ AE3 (d) as well as ACTH (e, ×200) confirming ACTH-secreting pancreatic neuroendocrine tumour. Ki-67 proliferative index was 5% with no mitotic figure seen (f).
Discussion/Conclusion
PNET is a heterogeneous neoplasm group with unique tumour biology, natural history, and prognosis compared to exocrine pancreatic tumours. Most PNETs are malignant in behaviour, with 60 % of them having metastases on pre-sentation [3]. These tumours are reported to have a dismal prognosis with 2-year and 5-year survival rates of 40 % and 16%, respectively [4]. Ectopic ACTH syndrome (EAS) is found in 5% to 10% of all cases of Cushing syndrome, and EAS caused by PNETs is considered rare yet aggressive [5]. This was evident in our case as the patient had liver metastases when she presented to us. One possible explanation for such phenomenon is that the metastatic lesions could have been the predominant source of ACTH secretion instead of the pancreatic lesion, leading to delayed clinical presentation [5]. However, there was no gallium uptake by the liver lesions in our case, but false-negative metastases have been reported in biopsy-proven NET, although gallium-68 scan is still an overall sensitive test in detecting small tumours with somatostatin receptor [6]. The ACTH-secreting PNET in this case was categorised as well-differentiated Pan NET G2 but had the florid presentation of hypercortisolism. This was attributed to the fact that there may be a dissociation between the severity of hypercortisolism and the tumour grades [7]. In addition, a clinicopathological study on ACTH-secreting PNET by Maragliano et al. found that most of the PNETs were of G1 and G2, although limited by small sample size [8].
Our literature review revealed female preponderance of EAS from PNETs as shown in this case and previous reports, although more studies are needed to confirm this possible association [1, 4, 5, 8, 9]. ACTH-secreting PNETs are more commonly found in the tail of the pancreas and the liver is the most frequent site of metastases. Clinical features of EAS from PNET include hypokalaemia, diabetes, lethargy, hypertension, and cushingoid facial appearance. Wu et al. reported a mean survival time of 23 months in patients with ACTH-secreting PNET [9]. The two treatment targets that need to be addressed in the management of EAS from PNET are control of symptomatic hormone oversecretion and prolonging survival by limiting tumour growth and burden. In general, hypercortisolism in EAS is considered as an endocrine emergency as mortality risk increases if left untreated, as illustrated in this case. The intensity of hypercortisolism should be aggressively reduced with pharmacological approaches or resection of the functioning tumour, and the decision rests on multidisciplinary team discussion tailored to the patient’s condition. Pharmacotherapies that inhibit steroidogenesis such as metyrapone and ketoconazole are the first-line treatment of hypercortisolism, and its efficacy is measured by normalisation of glycaemic control, hypertension, and hypokalaemia. Somatostatin analogues such as octreotide and lanreotide could be considered in EAS treatment, but their partial and transient effect in suppressing ACTH secretion has limited their use as monotherapy. Surgical excision of locoregional NET without distant metastases offers a cure for EAS [7]. In metastatic PNETs, especially those with liver involvement, cytoreductive surgery has a role in well-differentiated grade 1 or 2 NET, less than 50 percent hepatic replacement, surgically favourable anatomy, and normal liver function test. Radiofrequency ablation and hepatic artery embolization are the localised antitumor therapy directed to the liver metastases from PNETs. Bilateral adrenalectomy is the rescue treatment for severe EAS when inhibition of steroidogenesis with drugs has failed in unresectable PNET [7]. Systemic anti-neoplastic treatments for metastatic PNETs include everolimus, sunitinib, peptide receptor radionuclide therapy, capecitabine and temozolomide (CAPTEM) [3].
In conclusion, ACTH-secreting PNET is an aggressive disease that typically presents at an advanced stage with distant metastases. EAS from PNET is potentially fatal if left untreated, and a multidisciplinary approach should be adopted to manage these challenging cases. There seems to be a dissociation between the intensity of the hypercortisolism and the grade of NET in ACTH-secreting PNET. EUS-guided fine-needle aspiration or fine-needle biopsy should be considered in tissue acquisition to establish diagnosis of PNET and guide further therapies. Owing to the scarcity of cases reported and lack of consensus in management of EAS from PNET, more prospective data are needed to establish its best treatment strategy and recommendations.

