











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroduction
Liver cancer is the sixth cancer and the third cause of death from cancer worldwide, according to the 2020 GLOBOCAN report. The higher ranking of liver cancer in mortality over incidence reflects its dismal prognosis, with almost matching incidence and mortality rates [1].
Hepatocellular carcinoma (HCC) develops in the context of chronic liver disease (CLD), the main causes being B/C viral hepatitis, alcohol-associated liver disease (ALD), and nonalcoholic fatty liver disease (NAFLD). The epidemiology of CLD is changing and shaping the epidemiology of HCC. Hepatitis B (HBV)-associated HCC, which is still highly prevalent in Asia and Africa, is decreasing with the widespread HBV vaccination [2]. The breakthrough on hepatitis C (HCV) treatment is decreasing HCV-associated HCC. Instead, the obesity pandemic justifies the 5% increase in NAFLD prevalence in the last 5 years to 30% worldwide, even in regions classically with low prevalence such as Asia and East-North Africa [3]. The aggregate data result in NAFLD being the fastest-growing etiology for HCC [4, 5]. This review summarizes the available data on epidemiology of NAFLD-associated HCC and the rationale for its screening algorithms.
Epidemiology of NAFLD-Associated HCC
NAFLD accounts for 15% of HCC cases worldwide [6]. In the USA, the prevalence of NAFLD-attributable HCC in patients transplanted for HCC increased 8-fold: 2% in 2002 and 16% in 2016 [4]. Similarly, the prevalence of HCC in patients transplanted for NAFLD increased 12-fold (twice the increase in patients transplanted for HBV, three times the one in ALD, and 6 times in HCV) [4].
Regarding Europe, in France, NAFLD-attributable HCC increased from 2.6% in the period of 1995-1999 to 19.5% in 2010-2014, whereas HCV decreased from 43.6% to 19.5% [7]. In the UK, where historically the prevalence of HCV-attributable HCC is low, the mortality of HCC is increasing, with a particular rise in the referrals for NAFLD-associated HCC as compared to HCV- or ALD-associated HCC [8]. In the Newcastle-upon Tyne Hospitals NHS Foundation 2010 register, 35% of HCC cases were attributable to NAFLD [8].
In Latin America, NAFLD-attributable HCC in trans-planted patients increased 6-fold from 2005 to 2012 (from 2% to 13%), in contrast with only 1.5-fold increase in ALD and 2-fold decrease in HCV [5]. Regarding Asia, in Japan, nonviral etiology of HCC, including NAFLD and ALD, accounted for 10% of the cases in 1991, 24% in 2010 (representing 10% relative increase each year), and 32.5%in 2015 (representing an acceleration for 30% relative increase per year) [9]. One study from Japan described an even higher 46% NAFLD-attributable HCC, in 2018 [10]. Similar figures are observed in Korea, where NAFLD-attributable HCC increased from 3.8% during 2001-2005 to 12.2% in 2006-2010 [11].
Considering only cirrhotics, the trend sustains. A study with around 100,000 cirrhotic patients from the Veterans Administration healthcare system showed a 60% increase in the incidence of HCC in patients with cirrhosis diagnosed in 2008-2014, as compared to 2001-2007. The greatest increase, 88%, occurred in NAFLD-associated cirrhosis [12].
Meta-analyses showed an annual HCC incidence rate of 1.44% and 3.78% in patients with NAFLD-associated advanced liver fibrosis and cirrhosis, respectively, which seems similar to non-NAFLD-associated cirrhosis [13, 14], except for HCV-associated cirrhosis [12, 15]. The risk was 100 times lower in precirrhotic NAFLD: 0.03%per year [14]. Importantly, despite the low incidence of HCC in precirrhotic NAFLD, a retrospective study on around 300,000 NAFLD patients from the Veterans Health Administration showed an 8-fold higher risk for HCC compared with non-NAFLD controls, after adjusting to race and metabolic traits [16].
In noncirrhotic HCC, the proportion of NAFLD-attributable HCC is even higher. Indeed, the absence of cirrhosis is 5-fold more likely in NAFLD-associated HCC compared to non-NAFLD HCC [16].
NAFLD-associated HCC, compared to other etiologies, tends to occur in older patients, with more metabolic and cardiovascular comorbidities, and more frequently without liver cirrhosis (38% vs. 15%) [6]. NAFLD-associated HCC is more often detected outside specific surveillance [6], which could not be fully explained by the higher proportion of noncirrhotic HCC, since it also occurs in NAFLD-associated cirrhosis [17], even though HCC incidence is similar to other etiologies of cirrhosis [14, 15]. Indeed, HCC screening is less than half likely to be offered to a patient with NAFLD-associated cirrhosis as compared to HCV-associated cirrhosis, even though those patients do not comply less [18]. NAFLD-associated HCC tends to be larger, uninodular, with a more infiltrative pattern but more frequently well-differentiated, and with similar tumor stages at diagnosis [6]. Larger tumor size and uninodular involvement may be associated with the absence of cirrhosis, which confers lower resistance to expansive tumor growth [6]. Interestingly, despite overall survival in NAFLD-associated HCC patients seems similar compared to HCC from other causes [8], considering only cirrhotic patients, mortality from HCC seems 70% higher in NAFLD-associated cirrhosis [6], which may reflect treatment limitations due to comorbidities. Furthermore, preliminary data suggest that immunotherapy may be less efficient in NAFLD-associated HCC due to immune exhaustion and impaired immune-surveillance [19].
Risk Factors for NAFLD-Associated HCC
NAFLD-associated HCC occurs more frequently in noncirrhotic livers, as compared to other etiologies. A meta-analysis of 19 studies including over 150,000 patients showed that in precirrhotic CLD, NAFLD associates with 2.5-fold increased risk of developing HCC compared to other etiologies [20]. The presence of NASH further increases the risk for HCC [21]. Furthermore, 40% patients with NAFLD-associated HCC are precirrhotics, ranging from 10 to 75% among studies [16, 20]. Importantly, the prognosis of HCC depends on its stage and treatment approaches, whereas the presence of cirrhosis does not seem to independently associate with survival in NAFLD-associated HCC [22].
In NAFLD patients, the presence and severity of liver fibrosis are the most important risk factors for HCC development, whereas steatosis, ballooning, or lobular inflammation are not associated [23]. Despite the lack of association with histological inflammation, increased aminotransferase levels seem to increase the risk for cirrhosis and to double the risk for HCC [24]. High scores in noninvasive fibrosis tools, such as APRI, FIB-4, and NAFLD fibrosis score (NFS), are associated with an increased risk for HCC [23], even in the noncirrhotic range [10].
Liver stiffness measurement (LSM) may add in HCC risk stratification. While there is no clear LSM cutoff that accurately predicts the risk for HCC [25], there is a dose-dependent increase in the risk with higher LSM, in noncirrhotic and cirrhotic ranges [23]. For example, the 2-year risk was estimated as 9% if higher than 18 kPa and 13% if higher than 38 kPa [25]. The kinetics of LSM might also be predictive, as a retrospective study on 1,039 NAFLD patients showed that an increase in LSM was associated with a 70% increase in HCC risk, particularly for increases higher than 20% [26].
Older patients are more likely to develop HCC, even without cirrhosis, due to a defective cancer immune-surveillance and DNA damage with accumulation of gene mutations. Regarding metabolic dysfunction, type-2 diabetes mellitus (T2DM) is the strongest risk factor for HCC in NAFLD, increasing 3-fold the risk [16]. Smoking is another risk factor, with a synergic effect with T2DM. In cirrhotics, current smokers have a 50-80% increased HCC risk compared to never smokers [27].
Concerning alcohol, any alcohol consumption, even social intake (i.e., less than 3 drinks per day or 3-6 drinks daily on weekends), increases 3.5-fold the HCC risk in NAFLD-associated cirrhosis [15], whereas in noncirrhotic patients, the effect of moderate alcohol consumption remains controversial. However, we should take into consideration that alcohol and body mass index (BMI) present a synergic effect on the risk for HCC, with obese patients with mild alcohol consumption (defined as more than 3 drinks per week), presenting an almost 4-fold increased risk [28].
In NAFLD-associated cirrhotics, the known risk factors for HCC are male sex, older age (80% increased risk per decade), T2DM (2-4 times increased risk), decreased platelets count (translating portal hypertension), and low serum albumin (2 times increased risk) [23, 29-31]. Conversely, BMI, hypertension, or dyslipidemia does not seem to be risk factors, probably rendering the reversal of those metabolic factors as cirrhosis progresses [29]. Clinical decompensation further increases HCC risk in NAFLD-associated cirrhosis [30]. Also, even in compensated cirrhotics, 6 points on Child-Pugh-Turcotte score increase 2-fold the risk compared with 5 points [31].
Physiopathology of NAFLD-Associated HCC
European primary care healthcare records showed that, compared to the general population, NAFLD patients had a 3-fold higher chance of developing HCC. That risk increased by 8-folds in patients with steatohepatitis [21].
The increased risk for HCC in NAFLD may depend on the hepatic steatosis and liver injury per se, or on the metabolic conditions that led to hepatic steatosis, such as T2DM and obesity. Indeed, T2DM and/or obesity represented over one-third of the attributable fraction for HCC in the elderly, in a SEER-Medicare database [32]. Furthermore, in the general population, the number of metabolic abnormalities, such as T2DM, obesity, arterial hypertension, and dyslipidemia, has an additive effect on HCC risk [33].
Liver steatosis is, by itself, potentially carcinogenic, being a source of oxidative stress [34]. Oxidative stress induces DNA damage and the accumulation of methylated, hence silenced, tumor-suppressor genes. Accordingly, NASH patients with HCC present higher oxidative damage in hepatocytes [35]. Cholesterol and ceramides-related lipotoxicity may also favor carcinogenesis [34].
T2DM is potentially carcinogenic through different mechanisms: (a) activation of proinflammatory pathways and oxidative stress leading to genomic instability and apoptosis inhibition, (b) hyperinsulinemia and insulin-like growth factor [29], (c) T2DM-induced dysbiota and bacterial translocation, and (d) iron deposition. T2DM is consistently the strongest risk factor for HCC, in the general population [32], in patients with NAFLD [21, 33] and NAFLD-associated cirrhosis [29], and even in patients with non-NAFLD cirrhosis [36]. The effect of T2DM on HCC risk seems to be time-dependent, as T2DM duration less than 10 years is associated with a 3-fold increased risk, but duration longer than 10 years with a 5-fold increased risk as compared with the absence of T2DM [37]. Also, diabetic retinopathy seems a stronger predictor for HCC as compared to T2DM alone, which may translate a longer duration of T2DM or a disruption on VEGF physiology [38].
Epidemiological studies suggested that metformin was associated with 50% decreased risk for HCC [39], with a time-dependent effect: 7% decreased risk per year on metformin [40]. Reversely, insulin or sulfonylureas were associated with 2-fold and 50% increased risk, respectively [39]. Insulin may have carcinogenic effects promoting cell proliferation. Sulfonylureas promote insulin secretion potentially being indirectly carcinogenic. Accordingly, sulfonylureas’ potential carcinogenic effect seems to be restricted to first- and second-generation sulfonylureas, but not third-generation drugs such as glimepiride, which have lower insulin secretagogue effects [41]. Anti-HCC effects of metformin seem to be insulin-dependent and -independent: increases insulin sensitivity decreasing insulin levels, but also directly activates intracellular AMPK, leading to decreased protein synthesis and cell proliferation [40], decreases the stem cell population, and inhibits an epithelial-to-mesenchymal transition. The effect of thiazolidinediones is less clear, as a meta-analysis of 4 studies did not show a significant effect, even though all studies individually found a beneficial effect [39]. Thiazolidinediones present potentially anticancer effects by inducing cell growth arrest and apoptosis, while being able to inhibit cell invasion [42]. A study from Taiwan not included in that meta-analysis, on around half a million T2DM patients followed for 7 years, did find a dose- and time-dependent beneficial effect of both pioglita-zone and rosiglitazone on the risk of developing HCC [42].
Obesity seems to double the risk for HCC [43], accounting for 16% of its attributable risk [44]. There is dose-dependent increased risk for HCC according to the BMI category: 36% increase in overweight, 80% in class I obesity, and 300% in type II [45]. Even though lifestyle factors are difficult to dissect as risk factors for HCC, high physical activity seems to decrease in 25% the risk for HCC as compared to low physical activity [46].
Dyslipidemia is a risk factor for NAFLD/NASH, but its association with HCC is controversial [47]. However, consistent epidemiological data point out statins treatment to decrease 40-70% the risk for HCC [48], which may translate the pleiotropic effects of statins besides its cholesterol-lowering effect.
Genetic polymorphisms known to promote NAFLD have also been associated with increased risk for HCC. PNPLA3 (patatin-like phospholipase domain-containing-3) rs738409 variant impairs mobilization of triglycerides from lipid droplets and is associated with a 3-fold increased risk for HCC, independently of age, gender, BMI, presence of T2DM, or advanced fibrosis/cirrhosis [49]. TM6SF2 (transmembrane-6 superfamily member-2) rs58542926 variant hampers trafficking of pre-VLDL particles and is associated with a 2-fold increased risk for HCC when on homozygosity [50]. APOB (apolipoprotein B) variants are associated with increased HCC risk despite favorable lipid profile [51]. MBOAT7 (membrane-bound O-acyl transferase-7) ensures adequate cell membranes’ desaturation level by catalyzing the transfer of polyunsaturated fatty acids such as arachidonoyl-CoA to lysophosphatidylinositol. MBOAT7 rs641738 variant results in decreased MBOAT7 expression and is associated with a 2-fold increased HCC risk in NAFLD patients without advanced fibrosis [52].
Conversely, HSD17B13 (hydroxysteroid-17-beta dehydrogenase-13) rs72613567 variant is associated with a 40%protection for HCC [53].
Accuracy of Screening Methods for NAFLD-Associated HCC
Ultrasound (US) is the imaging modality recommended for HCC screening. However, US is operator-dependent, and 20% US examinations do not have adequate quality for the evaluation of hepatic lesions [54], rising to 33% in Child-Pugh-Turcotte class C patients, with BMI over 35 kg/m2, or with NAFLD-associated cirrhosis [55]. US detects HCC lesions with 84% sensitivity and 91% specificity; however, sensitivity drops to 47% for early HCC [56]. Interestingly, only 40% of HCC is detected at an early stage, and inadequate US sensitivity is the main reason for HCC being diagnosed at advanced stages during surveillance [57]. Patients with Child-Pugh-Turcotte class B/C cirrhosis have a 90% increased risk of inadequate US, due to lower accessibility of a severely atrophic liver retracted under the rib cage [55]. US performs particularly worse in NAFLD-associated cirrhosis, with an almost 3-fold increased risk of US diagnostic inadequacy [55]. Steatosis can impair visualization of deep liver nodules through increased attenuation of the US pulse [55]. To standardize the evaluation of the HCC-screening US accuracy, the American College of Radiology developed the Ultrasound Liver Imaging Reporting and Data System (US LI-RADS) algorithm that has 2 components: detection scores and visualization scores. The visualization score allows standardization of the US report regarding expected sensitivity, in 3 categories: A (no or minimal limitations), B (moderate limitations), and C (severe limitations to allow accurate visualization).
The diagnostic accuracies of CT and MRI are superior to US, particularly in patients with worse US LI-RADS visualization scores [58]. However, CT scans impose potential harms such as radiation exposure and contrast-induced nephropathy, and MRI is time consuming and costly. Recently, application of abbreviated MRI protocols to HCC screening presented higher sensitivity than US (82% vs. 53%), even though the performance declined for tumors smaller than 2 cm (69% vs. 86%) [59]. There are different abbreviated MRI protocols that consist of a limited number of sequences, not requiring contrast administration, hence allowing reduced acquisition and interpretation time [59].
Alpha-fetoprotein (AFP) is the best-studied HCC tumor marker, with the best cutoff, 20 ng/mL, presenting, in cirrhotics, a sensitivity of 41-61% at any stage (32-49%for early HCC) and specificity of 80-94% [60]. In combination with US, it increases 20% HCC-screening sensitivity versus US alone [56]. It can present false positives in cirrhotics particularly when aminotransferases are elevated, and in non-HCC malignancies such as cholangio-carcinoma and embryonal carcinoma of the testes. A different cutoff, 11 ng/mL, was proposed for non-HCV HCC [61]. Assessment should be longitudinal as increasing or fluctuating AFP levels might elicit intensive monitoring [62].
Lens culinaris agglutinin-reactive AFP (AFP-L3), desgamma-carboxy-prothrombin (DCP), and protein induced by vitamin K absence or antagonist-II (PIVKA-II) are other biomarkers. A small study on NAFLD showed PIVKA-II to present best accuracy (AUROC 0.86), while AFP-L3 lower (AUROC 0.689), compared to AFP (AUROC 0.763) [63]. DCP seems to outperform AFP in tumor recurrence after curative treatment in NAFLD (but not ALD)-associated HCC [64].
Clinical-laboratorial scores are being developed. GAL-AD score incorporates 5 parameters - sex, age, AFP, AFP-L3, and DCP - and seems promising in phase 2 studies for HCC screening [65]. GALAD score was validated in 2 NAFLD cohorts (from Germany and Japan), with better accuracy (AUROC >0.90) as compared to tumor markers alone, at any stage but particularly within the Milan criteria and in patients with precirrhotic NAFLD (AUROC 0.98). A GALAD score higher than −0.63 could be detected up to 1.5 years before the development of HCC [66] and hence could be used to stratify patients to intensive HCC-screening programs.
Novel tumor markers have been proposed, but need further validation. WTA+-M2BP (Wisteria floribunda agglutinin-positive Mac2-binding protein) or M2BPGI (Mac2-protein glycosylated isomer) is a noninvasive marker of liver fibrosis that seems promising as a NAFLD-associated HCC tumor marker [67]. M2BP is a secretory glycoprotein that, during fibrogenesis, undergoes modifications in N-glycosylation specifically recognized by WTA, resulting in WTA+-M2BP/M2BPGI. Among NAFLD patients with advanced fibrosis, the 1.25 cutoff presented 70% sensitivity, 78% specificity, and AUROC 0.806 for HCC. NAFLD patients with high versus low WTA+-M2BP/M2BPGI presented 21%versus 1.7% probability of HCC in 10 years [67]. ITIH4 (inter-alpha-trypsin inhibitor heavy chain-4) is upregulated by IL-6 and regulates the extracellular matrix. In preclinical and human NAFLD-associated HCC studies, higher ITIH4 levels are associated with NAFLD-attributable HCC, independently of liver fibrosis [68].
Lastly, AIM (apoptosis inhibitor of macrophages) is produced by macrophages and hepatic Kupffer cells and circulates in the bloodstream in two ways: inactive IgM-bound and active IgM-unbound, which helps in mediating the removal of excess fat, bacteria, cancer cells, and dead cell debris. In a small study with NAFLD patients, serum IgM-unbound AIM >1.6 μg/mL presented higher sensitivity for HCC detection than AFP and DCP (88.5% vs. 26.9% and 53.8%, respectively) [69]. Increased serum IgM-unbound AIM was detected up to 5 years before the diagnosis of NAFLD-associated HCC, which may represent a useful tool to select patients for HCC surveillance [70].
Current HCC-Screening Algorithms for NAFLD-Associated HCC
Cost-effectiveness of HCC screening in cirrhotics requires an annual incidence rate of at least 0.8-1.5%. In NAFLD-associated cirrhosis, similar to non-NAFLD cirrhosis, the HCC annual incidence rate is 1.5-4% [14, 15]; hence HCC screening is worthwhile. However, less than one-fourth of all patients with cirrhosis, and even less if NAFLD-associated cirrhosis, are enrolled in a HCC-screening program [57]. In the decision to enroll a patient in a HCC-screening program, other factors should be evaluated, such as age, overall health, functional status, willingness and ability to comply with screening, and eligibility for HCC treatment. International HCC-screening guidelines are consensual in supporting HCC screening for patients with NAFLD-associated cirrhosis (Table 1).
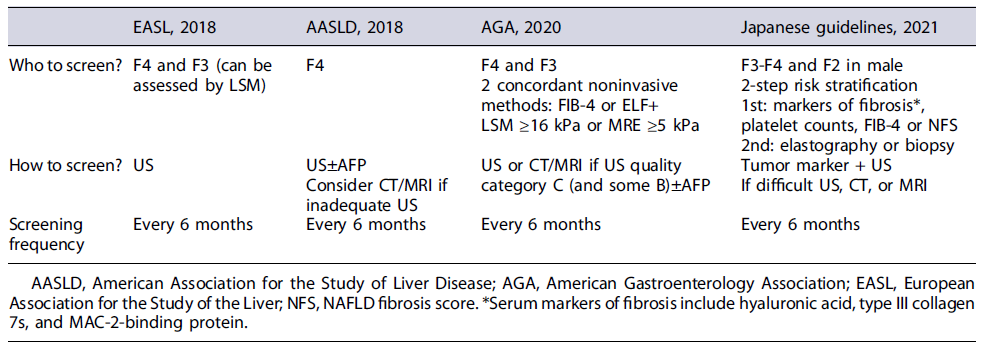
Cirrhosis can be assessed through noninvasive tools such as LSM. The Baveno Working Group proposed LSM higher than 15 kPa as the threshold for compensated advanced CLD and hence for starting HCC screening. Screening should be performed twice a year with AFP and US. NAFLD-associated cirrhotics with an inadequate US [55] might be offered other imaging modalities such as CT scan, MRI, or, more recently, abbreviated MRI [71], which will increase the screening costs. Ioannou et al. [72] proposed a risk-stratification model for patients with NAFLD-associated cirrhosis to optimize HCC screening. The model is available online (https://www.hccrisk.com) and integrates age, gender, BMI, T2DM, platelet count, AST, ALT, and albumin. It predicts a 3- and 5-year risk for HCC and categorizes in 3 risk groups: high risk with annual incidence >3%, medium risk 1-3%, and low risk <1%. Only patients with high or medium risk should be on HCC-screening programs, with the former probably benefiting from intensive strategies such as using abbreviated MRI [72].
Regarding precirrhotic NAFLD, guidelines do not recommend universal screening, since the annual risk of HCC development is 0.03-0.6% [14], lower than the accepted cost-effective 1.5%/year cutoff for cirrhotics. However, the 1.5% cutoff was delineated for cirrhotic patients, and cost-effective risk for HCC screening in noncirrhotic NAFLD patients could be lower. For example, in noncirrhotic HBV, an annual risk >0.2%/year is cost-effective [73], probably because those patients are eligible to different treatment modalities compared to cirrhotics, being able to tolerate large hepatectomy surgeries, and with higher life expectancy outside the tumor burden. Furthermore, because 6% of HCC occurs in precirrhotic NAFLD [6], an estimated global incidence of over 1 million cases by 2025 [74] results in 60,000 cases of HCC in noncirrhotic NAFLD that would be missed from HCC-screening programs each year.
HCC risk is heterogeneous among noncirrhotic NAFLD patients. For example in precirrhotic patients, high FIB-4 associates with 10 times higher HCC incidence compared to low FIB-4 [16]. Of note, the most recent guidelines already advise HCC screening in NAFLD patients with F3 fibrosis. An European cohort found that 90% of HCC in noncirrhotic NAFLD occurred in patients older than 65 years-old, suggesting that screening should not be considered in younger patients [17]. Preliminary evidence also suggests that noncirrhotic NAFLD patients 55 years old or older, with increased ALT and/or T2DM, are at particularly high risk [25]. Indeed, a study from Taiwan with a long-term follow-up of around 30,000 patients showed a 0.04% annual HCC incidence in young patients with normal ALT but 1.24%in older than 55 years with increased ALT [75]. Additionally, a population-based study from the USA suggested that noncirrhotic NAFLD patients that were male, older than 65 years old, smokers, diabetics, with increased ALT, had a 10-fold increased risk as compared to the whole noncirrhotic NAFLD patients, with a 0.45% annual incidence rate [30]. In NAFLD diabetic patients, the presence of diabetic retinopathy presented an AUROC 0.731 for HCC, being present in 15% of the diabetic population, but in 80% diabetic patients with HCC [38].
Two polygenic risk scores may help identifying high-risk NAFLD patients to guide a personalized HCC-screening program: (1) hepatic fat polygenic risk score (PRS-HFC) that combines known polymorphisms on PNPLA3, TM6SF2, GCKR, and MBOAT-7; and (2) polygenic risk score 5 (PRS-5) adjusted to HSD17B13 splice variant [76]. Both performed similarly: PRS-HFC ≥0.532 and PRS-5 ≥0.495 associated with 15 times higher HCC risk in the general population, 3 times in NAFLD patients (2 times in noncirrhotic patients), 5 times in obese, and 4.5 times in T2DM patients. Those scores presented very high specificity of 90%, but sensitivity below 30%. As such, those scores may help selecting patients to HCC screening, but should not be used to exclude patients from HCC screening.
A 133-gene signature in the hepatic transcriptome was proposed as a prognostic liver signature in NAFLD patients (PLS-NAFLD), which stratifies patients in high risk (23%15 years HCC rate) and low risk (15 years HCC rate of 0)[77]. This score performs well even in patients with minimal liver fibrosis. Interestingly, high-risk genes were associated with immune cells activation, induction of tolerogenic IDO1+-dendritic cells and T-cell exhaustion (a known feature of NAFLD-associated carcinogenesis [19]), whereas low-risk genes were associated with FXR and FGF-19/21 pathways. Furthermore, a 4-protein secretome signature (PLSec-NAFLD) also stratified in high risk for HCC (38%15-years incidence) and low risk (none developing HCC)[77]. High-risk proteins were lymphoprotectin and progranulin, and low-risk proteins angiopoietin-2 and hepatocyte growth factor receptor. These scores might help excluding patients from HCC screening.
Extrahepatic cancer screening programs should also be encouraged to NAFLD patients, since extrahepatic cancers are 8 times more frequent than HCC and not associated with liver fibrosis [13]. A meta-analysis showed an annual incidence rate of 1.16%, being the most frequent ones uterine, breast, prostate, colorectal, and lung cancers [13].
Conclusion
A HCC-screening program requires 3 premises: (1) the population on surveillance must have high risk for HCC;(2) screening tools must be effective in diagnosing treatable HCC in that population; and (3) those patients must be eligible for effective treatments with expected impact on survival and/or quality of life.
Regarding the first premise, patients with NAFLD-associated cirrhosis have a HCC incidence rate superior to the cost-effective 1.5% cutoff and should undergo HCC screening. However, 20-50% of NAFLD-associated HCC occurs in patients with precirrhotic disease. The actual cost-effective cutoff in this population is yet to be determined. Furthermore, the risk of HCC in NAFLD patients is highly variable, being only significant in older patients, and screening should not be proposed in younger than 65 years old. Research is moving toward precision-medicine, and tools to thoroughly stratify patients are in development. One example is polygenic risk scores, which showed promising results.
The second premise, effective screening tools, has some specific limitations in NAFLD patients. Indeed, the accuracy of US decreases in obese and patients with liver steatosis, rendering usual screening programs less accurate for NAFLD patients. Abbreviated MRI is appealing for patients with unreliable US.
The third premise, effective HCC treatment, also has its caveats in patients with NAFLD-associated HCC, which tend to be older and with comorbidities that may limit treatment options, as well as preliminary data showing less efficacy of first-line systemic therapies in NAFLD-associated HCC. HCC screening in patients with NAFLD is challenging but should be improved, so we can change current statistics in which NAFLD is the underlying etiology with the highest proportion of HCC diagnosed outside screening programs.

