











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroduction
Erythema elevatum diutinum (EED) is an uncommon variant of chronic leukocytoclastic vasculitis that presents as red, violaceous, or red-brown papules, plaques, and nodules, typically in a symmetric fashion on the extensor surfaces of the limbs, with a preference for the hands, feet, elbows, knees, and Achilles tendons1-3. Lesions of EED are usually asymptomatic but can be associated with pruritus, pain, or stinging4. Less commonly can also present with extracutaneous manifestations, including joint pain, constitutional symptoms, and ocular involvement5.
It can affect individuals of any age but is most common in adults between the ages of 40 and 65, apparently without preference for sex or race1,6.
EED pathogenesis remains poorly understood, but it is believed to be a consequence of immune complex deposition in dermal blood vessel walls, resulting in complement activation, with selective chemotaxis of neutrophils, through cytokine activation (specifically interleukin-8), and the release of destructive enzymes. This repetitive vascular damage ultimately progresses to fibrosis. Pathogenic involvement of antineutrophil cytoplasmic antibodies is also possible2.
EED can be associated with various conditions, including hematological malignancies (Immunoglobulin A [IgA] monoclonal gammopathy, paraproteinemia, myelodysplastic syndromes, myeloproliferative disorders, hairy cell leukemia), infections (streptococcal, hepatitis B and C, HIV, syphilis, tuberculosis), inflammatory/autoimmune disorders (inflammatory bowel disease, granulomatosis with polyangiitis, cryoglobulinemia, relapsing polychondritis, celiac disease, systemic lupus erythematosus, and rheumatoid arthritis), and solid tumors (breast and lung cancer)7,8.
Additionally, Momen et al.,’s review9 spanning from 1977 to 2012 revealed EED’s association with numerous other disease processes, including HIV, IgA gammopathy, bacterial infections, and inflammatory bowel disease, among others. More recently, Doktor et al. in their 2019 review2 of 133 cases of EED between 1990 and 2014, identified 21 cases associated with HIV6.
Clinical case
We describe the case of a 41-year-old Caucasian male with no significant personal or family medical history, referred to the dermatology consultation by his primary care physician due to the presence of nodular lesions in the lower third of the legs and ankles, bilaterally. These lesions began to appear approximately 1 year before as erythematous maculopapular lesions and over time, they become more raised and nodular. They were asymptomatic and there was not any associated systemic symptom.
The clinical examination revealed bilateral red papules, nodules, and areas of hyperpigmentation in the patient’s legs and ankles. Lesions were moderately firm and mobile, with the plantar surface of the feet remaining intact (Fig. 1).
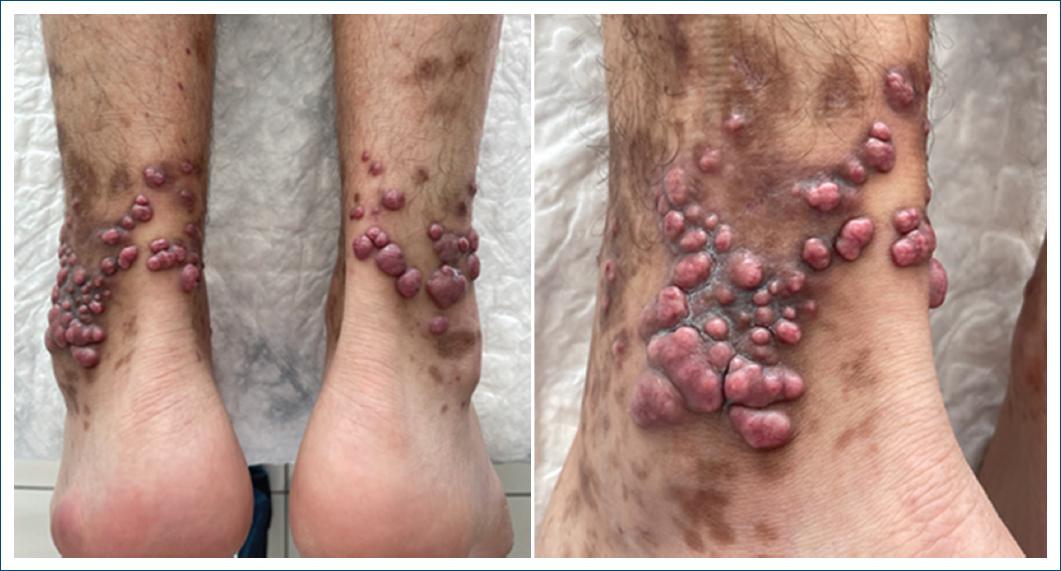
Figure 1 Bilateral red papules and nodules along with areas of hyperpigmentation on the legs and ankles.
A skin biopsy was performed on a nodule, revealing features consistent with concentric fibrosing vasculitis with a neutrophilic infiltrate and leukocytoclasia (Fig. 2).
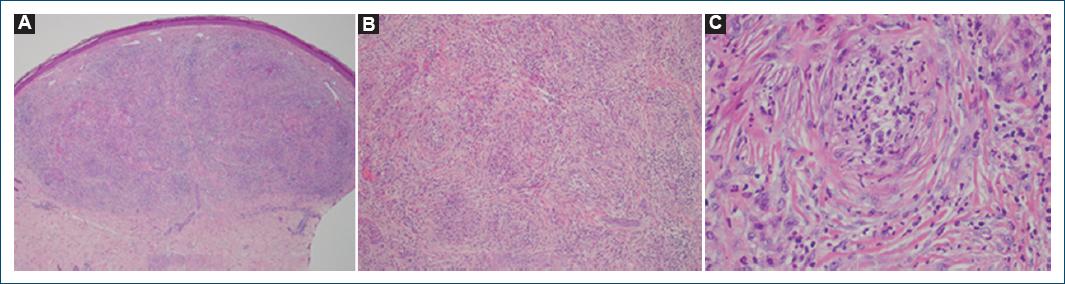
Figure 2 Fibrosing concentric vasculitis with dermal neutrophilic cell infiltrate and leukocytoclasia can be clearly observed at high magnifications. A: magnification ×40. B: magnification ×100, C: magnification ×400: hematoxylin and eosin.
Based on the clinical and histopathological findings, the diagnosis of EED was established.
An extensive panel of serological, immunological (including Immunoglobulin G antineutrophil cytoplasmic antibodies), and sexually transmitted infection screening tests was conducted, revealing positive results for HIV-1: 40,000 copies/mL and a CD4+ cell count of 387 cells/mcL; and Neisseria gonorrheaedetected by polymerase chain reaction in urine. Chest X-ray and glucose-6-phosphate dehydrogenase levels were normal and there were no significant immunological, hematological, or renal abnormalities detected.
Considering the diagnosis of EED, treatment was initiated with oral dapsone at a daily dose of 100 mg.
In addition, a single dose of intramuscular ceftriaxone 500 mg was administered to treat Neisseria gonorrhea infection.
After 7 months of dapsone treatment, there was an almost complete regression of the lesions (Fig. 3) and the dose was reduced to 50 mg/day, being discontinued after 12 months, when the lesions had completely regressed, leaving only residual hyperpigmentation (Fig. 4). Throughout the treatment course, no clinical or hematological side effects were observed.
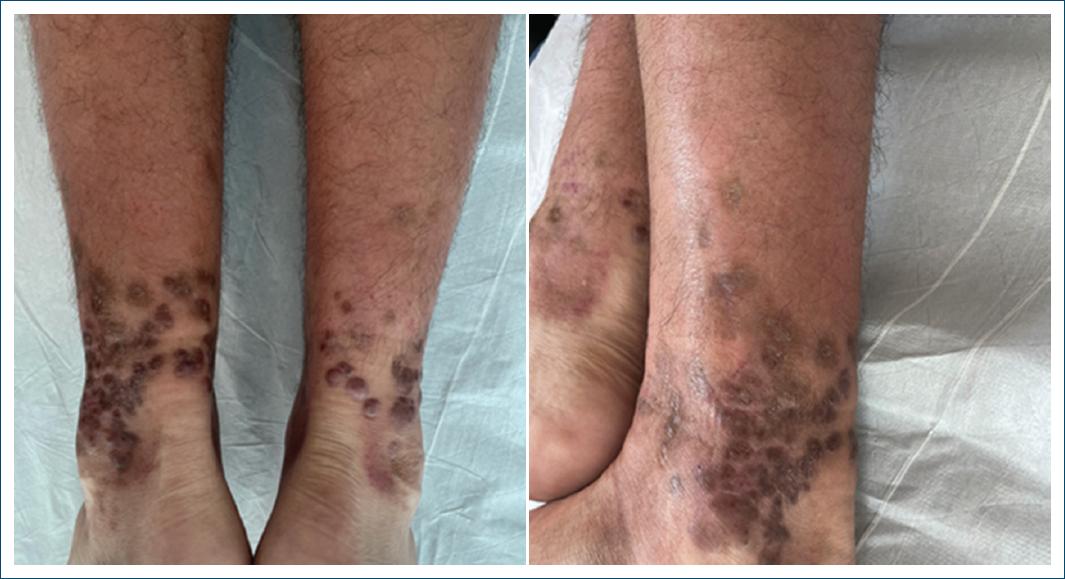
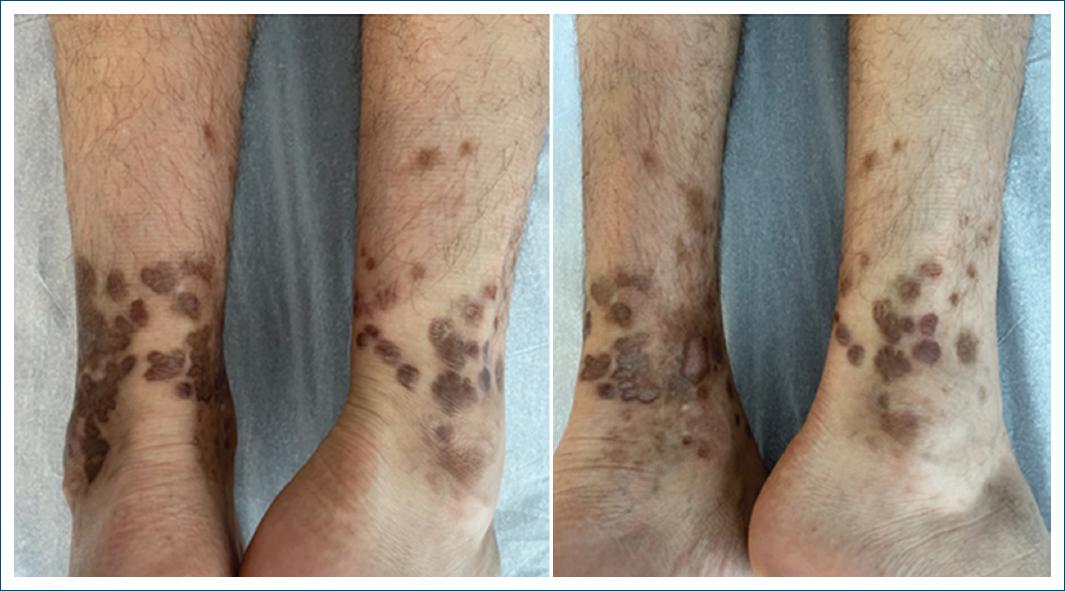
Figure 4 Complete regression of the lesions, with only residual hyperpigmentation remaining, after 12 months of treatment.
The patient was also referred to an infectious diseases consultation and antiretroviral therapy with lamivudine and dolutegravir was started a few months later (approximately when dapsone was reduced), with no detectable viral copies in the follow-up study at 3 months.
Discussion
EED is a rare, chronic leukocytoclastic vasculitis characterized by a typical eruption pattern of bilateral and symmetrical papules, nodules, and plaques over the extensor surfaces of the body, such as elbows, knees, and ankles1,2. Face, ears, and mucosal membranes are usually spared3. Initially, lesions are soft and display a reddish or purplish hue, but over time, they can become red-brown or yellow-brown and firmer due to fibrosis4. Hemorrhage and ulceration are not common, often resulting in subsequent hyperpigmentation on regression. The nodular form is rarer and generally occurs in patients with HIV infection, just like in our patient6.
The pathophysiology of EED in the context of HIV infection is multifaceted, involving immune dysregulation, chronic inflammation, and deposition of immune complexes in the small blood vessels of the skin. This immune complex deposition triggers a cascade of inflammatory responses, including the recruitment of neutrophils and other inflammatory cells, leading to vessel wall damage and subsequent fibrosis. The mechanism involves, therefore, a complex interplay between immune dysregulation, chronic inflammation, and impaired clearance of immune complexes6.
Despite reports where spontaneous resolution occurs within a span of 5-10 years, the disorder generally follows a persistent course over years, hence the word diutinum (meaning persistent or long-lasting) in the name of the disease. Individual lesions may regress (with hyperpigmentation) as new ones continue to appear8.
The diagnosis of EED is based on clinical and histopathological findings, with histology being not specific but suggestive and essential to establish a diagnosis.
Histologically, during the initial phases, the condition displays leukocytoclastic vasculitis with the presence of polymorphonuclear cells, macrophages, and histiocytes within the dermal layers. As the disease progresses to its later stages, the infiltrate evolves to encompass features, such as granulation tissue, fibrosis, vascular proliferation, a lymphohistiocytic inflammatory infiltrate, and residual foci of leukocytoclastic vasculitis6.
The clinical differential diagnosis of EED includes Sarcoidosis, Sweet’s syndrome, neutrophilic dermatosis of the hands, rheumatoid neutrophilic dermatosis, palisaded and granulomatous neutrophilic dermatosis, and granuloma annular. In late-stage disease, the differential diagnosis encompasses mainly dermatofibroma, tuberous xanthomas, rheumatoid nodules, and multicentric reticulohistiocytosis3.
In our case, the diagnosis was suggested by the histopathological findings along with a negative immunologic panel.
The standard and most effective treatment for EED is dapsone, typically administrated in doses ranging from 50 to 150 mg daily2. Other treatments, such as colchicine, tetracyclines, topical betamethasone, niacinamide, and systemic corticosteroids (e.g., prednisolone) have also been reported6,8. Here, due to a strong clinical suspicion of EED before the confirmation of HIV, dapsone was immediately started. Despite the underlying immune dysregulation in HIV, dapsone’s multifaceted anti-inflammatory mechanisms effectively control the chronic inflammation and immune complex deposition characteristic of EED.
The triggers and the associated disorders must be treated as well. For example, in patients with HIV, the usual approach involves integrating antiretroviral therapy into the treatment regimen7,8 which was begun a few months later (mostly due to a delay in obtaining the test results and the missing of several appointments). Despite this in, our patient’s case, a favorable response to dapsone was observed. Furthermore, there have been no recurrent lesions observed following cessation of this drug (with a follow-up period of 7 months to date).
Conclusion
This case report aims to raise awareness about the potential occurrence of EED and its serious associated conditions, including hematological malignancies, solid tumors, autoimmune disorders, and infectious diseases, such as HIV, HCV, and HBV8. A comprehensive investigation into its cause is crucial to exclude other associated conditions, given the diverse and frequently severe underlying factors that can pose life-threatening risks.

