











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroduction
Hereditary palmoplantar keratodermas (PPK) are a heterogeneous group of rare skin disorders characterized by thickening of the epidermis of palms and soles1. PPK can be classified by the pattern of lesions into four clinical subtypes: diffuse, punctate, focal, and striate1,2. Isolated punctate PPK can be further subdivided into three variants: punctate PPK type 1 (Buschke-Fischer-Brauer disease), punctate porokeratosis (type 2), and acrokeratoelastoidosis (type 3)3. Herein, we describe the clinical and genetic features of two family members with punctate palmoplantar keratoderma type I.
Clinical case
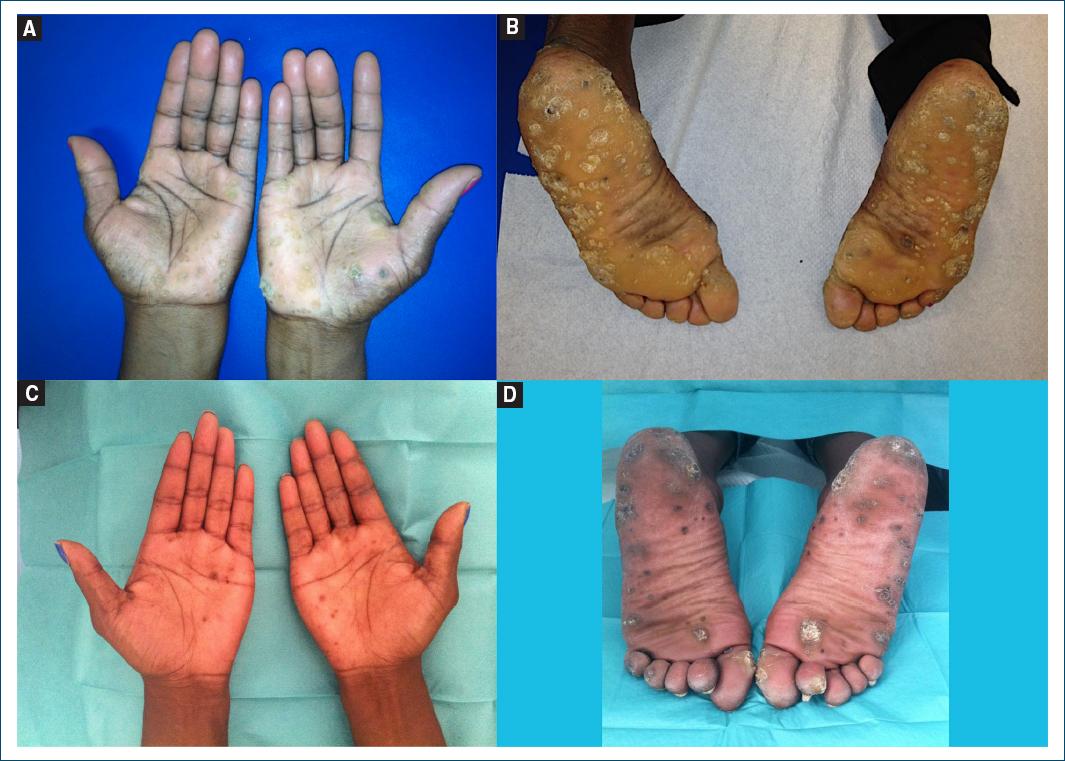
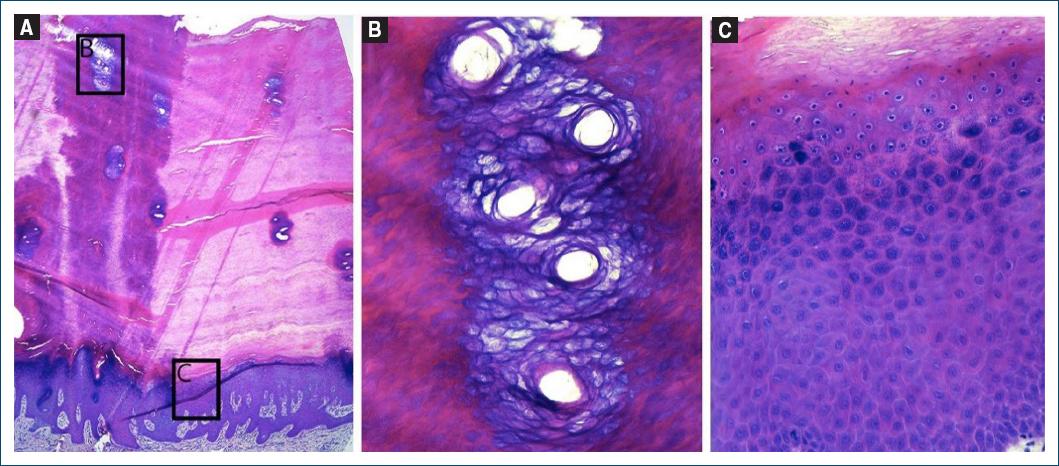
A 61-year-old African woman presented with a 10-year history of multiple, small (0.3-1 cm), circular hyperkeratotic papules and plaques on the palms and soles (Figs. 1A and B). These skin abnormalities were asymptomatic and irregularly distributed. There were no additional complaints, such as keratoderma transgrediens, and no history of arsenic exposure, immunosuppression, or malignancy. Notably, her 29-year-old daughter also exhibited a similar dermatosis (Figs. 1 C and D). A skin biopsy revealed marked orthohyperkeratosis overlying acanthotic epidermis with elongated and curved rete ridges (Fig. 2A) with prominent, coiled and dilated acrosyringia (Fig. 2B) and hypergranulosis, with no cytologic features of HPV infection (Fig. 2C). A computed tomography body scan ruled out malignancy. Based on the clinical and histological features, a diagnosis of type I hereditary punctate keratoderma was established. Genetic study of the AAGAB gene in both the mother and daughter identifies a heterozygous mutation in intron 5: c.535+1G>A. Subsequent mRNA analysis confirmed that the splicing mutation led to the deletion of exon 5, resulting in decreased protein levels. Notably, the mother reported that her sister and nephew had similar manifestations. As they both live abroad, they did not undergo genetic testing. The patients were treated with salicylic acid 30%, urea 40%, and tretinoin; however, only minimal clinical improvement was observed in both cases. Acitretin treatment was proposed to the mother, but the patient ultimately did not proceed with it. Over 5 years of follow-up, neither patient developed a history of neoplasia.
Discussion
Punctate palmoplantar keratoderma type I (PPPK1), first described in 1910, is a rare autosomal dominant inherited disorder characterized by multiple tiny punctate keratoses on the palms and soles2. Its estimated prevalence is approximately 1.17/100,000 individuals4. Lesions typically begin to develop in early adolescence but can also manifest later in life, as late as the fifth decade, as observed in our patient. PPPK1 is distinguished by numerous pinpoint, firm papules, 2-8 mm in diameter, which may progress to become translucent, opaque, or verrucous over time4. The papules gradually increase in number and size with age, and often coalesce in pressure-bearing areas of plantar skin2. The exact etiology of PPPK1 is not fully understood, but it is believed to involve a combination of genetic and environmental factors3,4. Several loci have been reported in the literature, with AAGAB being considered a major genetic factor3. This gene encodes the alpha- and gamma-adaptin binding protein, also known as p34. Mutations in this gene can increase epidermal growth factor receptor protein expression and tyrosine phosphorylation, resulting in cellular hyperproliferation2,5. To date, at least 50 different AAGAB mutations have been reported1, including the c.535+1G>A mutation identified in our patient, which has been previously documented only once in a sporadic case within the Chinese population6. In addition, not all patients with a PPPK1 phenotype have an identified variant in AAGAB, suggesting the potential involvement of other, yet-to-be-identified causative genes3. Histological examination of PPPK1 typically reveals hyperkeratosis, unspecific changes such as acanthosis, parakeratosis (in some cases) and hypergranulosis. The dermis is usually normal and devoid of inflammatory infiltrate4. Clinically, PPPK1 can exhibit varying degrees of severity between families, though the genotype-phenotype correlation remains poorly defined2. In the cases described, both the mother and daughter exhibited similar clinical severity, despite their symptoms starting 20 years apart. Although there have been reports suggesting an association between PPPK1 and malignancy, this link remains somewhat controversial3. Differential diagnoses to consider include verruca vulgaris, arsenic keratosis, punctate porokeratosis, acrokeratoelastoidosis, and focal acral hyperkeratosis4. The treatment for PPPK1 is primarily symptomatic, and typically involves topical keratolytics such as lactic acid, urea, and salicylic acid. Additional treatment options include phototherapy, systemic retinoids (such as acitretin and alitretinoin), and surgical interventions. These therapies generally lead to temporary reductions in skin thickness and improvement in skin softness4.
Conclusion
This case report highlights the clinical, histological, and genetic findings of two family members with PPPK1. The clinical similarity between mother and daughter, despite differing ages of onset, illustrates the variable expressivity of the condition. Histopathological and genetic analyses played a key role in confirming the diagnosis. Continued research into the molecular mechanisms of PPPK1 is essential to idevelop more effective therapies.

