











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
INTRODUCTION
IgG4‑related disease (IgG4‑RD) is a systemic, immune mediated disorder typically characterized by fibroinflammatory lesions that can affect almost any organ.1‑3
IgG4‑RD was identified as a distinct systemic disease only a decade ago and knowledge about this condition is still growing.4,5IgG4‑RD can affect any organ, either concurrently or sequentially. It often manifests as a multiorgan disorder (60%‑90%) making it easily confounded with malignancies, infections, or other immune conditions. Most commonly affected organs include the pancreas (20%-60%), major salivary glands (submandibular, parotid, sublingual), orbits, lacrimal glands, biliary tree, lungs, kidneys, aorta, retroperitoneum, meninges, and thyroid gland (Riedel’s thyroiditis). Inflammatory lesions can form tumefactive masses, potentially leading to organ destruction.1,2
Clinical manifestation depends on the type of organs involved. Usually it presents subacutely, lymphadenopathy is common, and asthma or allergy are present in 40% of patients.1 The disease incidence peaks between the fifth and seventh decade with a male predominance (61%-80%).1,2The precise aetiology of IgG4‑RD remains elusive. Environmental triggers have been implicated, but clear genetic associations remain undefined. B and T cells are acknowledged as central to its pathogenesis, but their specific roles are not fully understood.1,2,6Although many of the pathophysiologic details of this condition remain to be elucidated, the pathology is essential to the diagnosis.1,7Essential pathological features encompass dense lymphoplasmacytic infiltrates, storiform fibrosis, and obliterative phlebitis. Immunohistochemical confirmation with IgG4 staining is crucial, with IgG4‑positive cells prevailing over other immunoglobulins. A diffuse plasma‑cell infiltrate with more than 10 to 200 IgG4‑positive cells per high‑power field and an IgG4 to IgG ratio exceeding 40%‑50% in a plasma‑cell infiltrate is indicative of IgG4‑RD.1,2,8,9Despite most patients present elevated serum IgG4 levels, 30%‑50% may exhibit standard levels with characteristic histopathological findings. Besides serum IgG4 elevation, an increase in other IgG subclasses is frequent, and contributes to the hypergammaglobulinemia often observed in IgG4‑RD patients. IgE elevation and mild eosinophilia are common. ANAs can be found at low titre, while anti‑ENA and anti‑dsDNA antibodies are usually negative. Elevated concentrations of IgG4 in tissue and serum are helpful in diagnosing IgG4‑RD, but neither one is a specific diagnostic marker. Histology and immunohistochemistry are the gold standard to achieve the diagnosis.1,2Renal involvement is observed in 7%‑24% of cases and may occur both directly (IgG4‑related kidney disease, IgG4‑RKD) or indirectly, as a consequence of post renal ureteral obstruction due to retroperitoneal fibrosis (IgG4‑RD RF).2,3The most frequent presentation of IgG4‑RKD is IgG4‑related tubulointerstitial nephritis (TIN), either as a mass‑like lesion or renal failure or even both and can be associated with declining renal function or rapidly progressive renal insufficiency and sub‑nephrotic proteinuria.3,4In IgG4‑related TIN, differently from other forms of TIN (drug‑induced or associated with systemic autoimmune disorders as Sjogren’s syndrome), the involvement of renal parenchyma is not diffuse but rather characterized by focal lesions surrounded by normal tissue.3,10Glomerular disease can be present mostly as membranous nephropathy (MN), and less frequently as IgA nephropathy and membranoproliferative glomerulonephritis. MN represents 7% of all IgG4‑RKD.3,4
Differential diagnosis between coexistent primary MN or secondary MN to IgG4‑RD “IgG4‑related MN” is supported by the simultaneous presence of TIN, the detection of other localizations of the disease (the kidney is very rarely the only organ affected), and the absence of antibodies specific for phospholipase A2 receptor (anti‑PLA2R) helps to support the diagnosis (American College of Rheumatology (ACR) and European League Against Rheumatism (EU‑ LAR) IgG4‑RD classification criteria consider anti‑PLA2R as an exclusion criterion for the diagnosis of IgG4‑RD).3,7,8IgG4‑related MN may occur with or without IgG4‑related TIN, and must be considered in the presence of proteinuria in patients with IgG4 TIN on biopsy.4,6,7,11,12
The comprehensive diagnosis of IgG4‑RD can be challenging and correlation among clinical, serologic, radiologic, and pathologic data is required for diagnosis.1 In 2019, the ACR and the EULAR developed a 3‑step classification criteria for IgG4‑RD that include involvement of at least one of 11 possible organs, exclusion criteria consisting of a total of 32 clinical, serologic, radiologic, and pathologic items and weighted inclusion criteria domains, addressing clinical findings, serologic results, radiology assessments, and pathology interpretations. ACR/EULAR criteria for IgG4‑RD could diagnose most patients with IgG4‑RKD as having IgG4‑RD.8,12Specific diagnostic criteria for IgG4‑RKD was developed in 2011 and revised in 2020 for the Japanese population, with sufficient specificity (90.0%), but relatively low sensitivity (72.7%).9,12,13For patients with affected vital organs, timely intervention is imperative to avert severe organ dysfunction. Glucocorticoids, initiated at 0.4‑0.6 mg/kg/day of prednisolone or prednisone, are the typical first‑line treatment. Subsequent therapy includes tapering over three to six months, followed by maintenance for up to three years. Immunosuppressants like azathioprine and mycophenolate mofetil were used as glucocorticoid‑sparing agents or remission‑maintenance drugs after glucocorticoid induced remissions, but their efficacy has never been tested in clinical trials. Furthermore, rituximab is considered for refractory cases.1‑3
CASE REPORT
An 87‑year‑old Caucasian male presented to the hospital with a two‑month history of fatigue with marked limitation of physical activity and generalized oedema. Patient denied changes in urine characteristics and output. His past medical history includes hypertension, dyslipidaemia, ischemic cardiomyopathy, and chronic obstructive pulmonary disease (COPD). Baseline serum creatinine level was 1.2‑1.6 mg/dL.
On physical examination the patient had a blood pressure of 241/129 mmHg, heart rate of 92 bpm, a 4+ bilateral lower extremity oedema, upper extremity, and lumbar region. Lung auscultation revealed decreased vesicular murmur in the right base. The remaining clinical examination was unremarkable. At admission laboratory results showed renal dysfunction with creatinine of 3.29 mg/dL, urea of 119 mg/dL, no ionic imbalances, and urinalysis showed hematoproteinuria with a P/C ratio of 11g/g, without evidence of dysmorphic erythrocytes. The patient was admitted for further investigation of nephrotic syndrome.
Blood tests revealed normochromic normocytic anaemia [Hb 9.5 g/dL with no vitamin or iron deficiencies, hypoalbuminemia [1.41 g/dL , normal peripheral blood smear, no monoclonal gammopathy on serum electrophoresis and immunoglobulins (IgA, IgM, IgG) within normal values (only a slightly increased IgA).
Immunologic study showed no hypocomplementemia (C3 140 mg/dL [90‑140 and C4 54 mg/dL [10‑40 ) and anti‑nuclear antibodies (ANA), anti‑double stranded DNA (dsDNA), anti‑neutrophil cytoplasmic antibodies (ANCA) and phospholipase A2 receptor (PLA2r) antibodies were negative. HIV, hepatitis B and hepatitis C serologies were also negative. The remaining analytic results are presented in Table 1.
Kidney’s ultrasound showed preserved form and dimensions, reduced corticomedullary differentiation and renal cysts, as well as non‑obstructive component. Bladder ultrasound was normal. Cardiac ultrasound showed concentric left ventricular hypertrophy with normal ejection fraction (55%‑65%), good global systolic function of the right ventricle. To exclude the presence of other conditions known to be associated with the nephrotic syndrome, a thorax, abdomen, and pelvic computed tomography and a colonoscopy were performed, both without abnormal findings.
Kidney biopsy was performed (left kidney) and anti‑proteinuric treatment and anticoagulation were initiated while admitted. After 20 days in hospital and 10 days of anti‑proteinuric treatment the patient was discharged with stabilized renal function (creatinine of 4 mg/dL) waiting for the final report of the kidney biopsy (primary suspicion was membranous nephropathy associated with chronic interstitial nephritis). Kidney biopsy findings were consistent with membranous nephropathy and tubulointerstitial nephritis (Fig. 1).
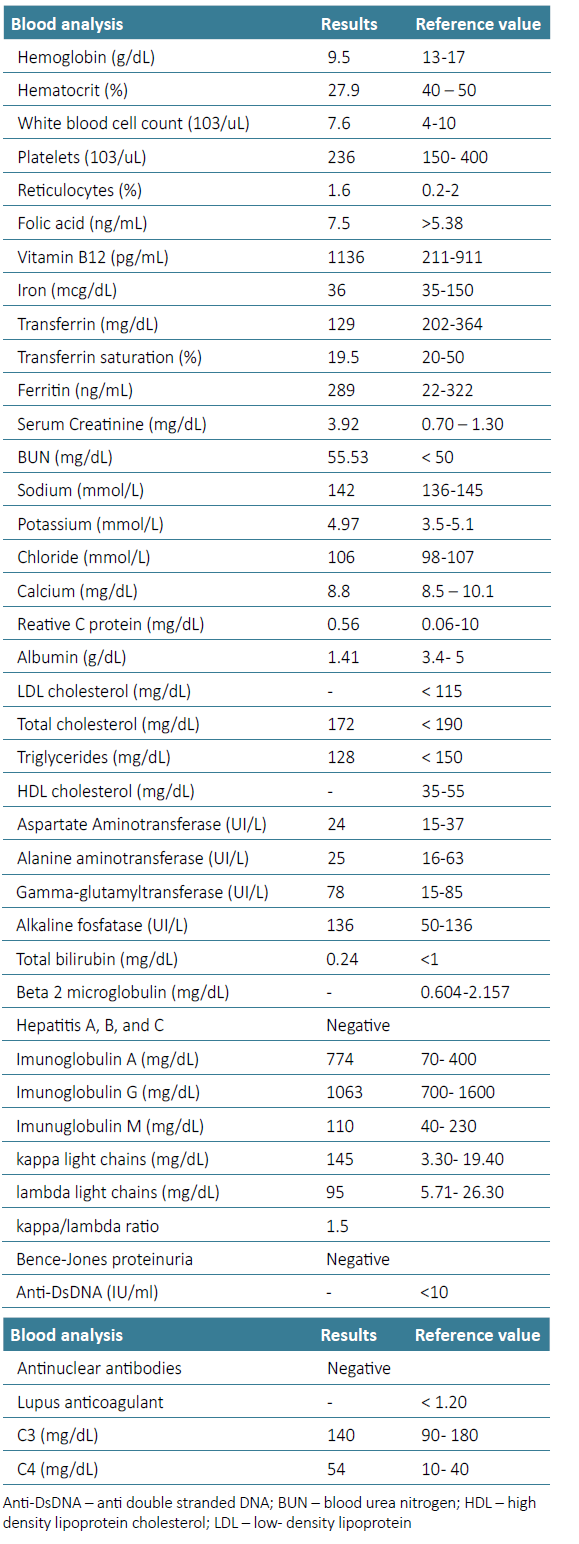
Figure 1. Kidney Biopsy‑ Histopathological Features (A): Light microscopy PAS staining‑capillary wall thickening and segmental endocapillary hypercellularity (20x). (B): Intense inflammatory infiltrate in the middle cortical region, occupying around 60% of the sample, consisting of mononucleated cells (200x). (C): Inflammatory infiltrate with tubular destruction and interstitial fibrosis (“storiform fibrosis”) already established. Foci of acute tubular necrosis and tubular destruction in the infiltrate area (20x). (D): Immunofluorescence: Parietal granular deposits of IgG‑κ and IgG‑λ (50x). (E) & (F): Immunohistochemistry: Lymphocytes and plasma cells (> 10 IgG4 plasma cells/hpf) and IgG4 deposits in the glomerular basement membrane (20x).
The sample submitted for light microscopy contained seven glomeruli, of which one was globally sclerosed. Non sclerosed glomeruli showed capillary wall thickening and segmental endocapillary hypercellularity with mononuclear cells. As well as diffuse podocyte hypertrophy. The interstitial fibrosis showed a storiform pattern and extended to 60% of the renal parenchyma. Foci of acute tubular necrosis and tubular destruction were present within the areas of interstitial inflammation. Immunofluorescence microscopy showed parietal granular deposits staining for IgG (2+), k light chains (2+), and λ light chains (1+/2+) along glomerular capillary walls. Immunohistochemistry showed inflammatory infiltrates consisted of lymphocytes and plasma cells, with more than 10 IgG4 plasma cells/ hpf being observed. IgG4 deposits were also observed in the glomerular basement membrane (Fig. 2). There was no evidence of changes in arterioles.
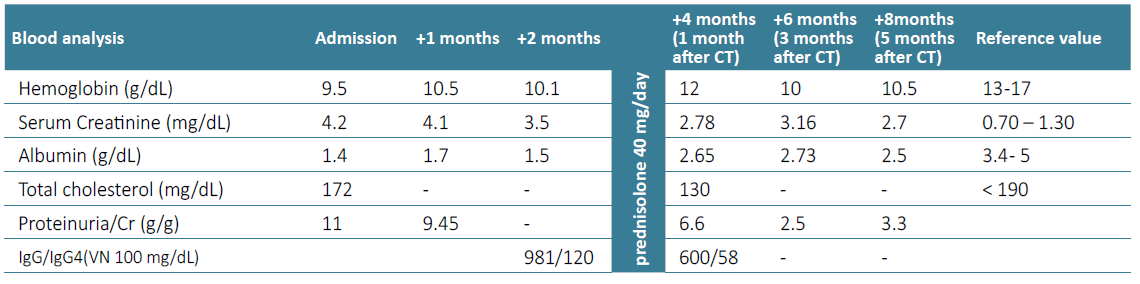
Considering those findings, subclasses of IgG were measured in the blood with an increase, albeit slight, of IgG4 (115 mg/dL, normal value 9‑100 mg/dL). Further exams excluded other organ involvement, namely pancreas, retroperitoneum, pleura, lung parenchyma. Thus, the clinic, histology, and the elevation of serum IgG4, although slight, supported the diagnosis of IgG4 disease with glomerular (membranous) and interstitial involvement.8 Furthermore, workup for circulating anti‑PLA2R anti‑bodies was negative, suggesting that the membranous nephropathy was not “primary” and maybe was linked to the IgG4‑related disease. Prednisolone was started at 40 mg/day. After one month of treatment nephrotic syndrome was ameliorated and his renal function improved, with a reduction of creatinine, from 4 mg/dL to 2.7 mg/dL, and proteinuria from P/C ratio 11 g/g to 2 g/g. Additionally improvement in anaemia (from 10 g/dL to 12 g/dL) and hypoalbuminemia (from 1.8 g/dL to 2.8 g/dL) occurred. The patient has been closely followed‑up to date (Table 2).
DISCUSSION AND CONCLUSION
IgG4‑related disease has been noted in many organs, including the kidney, resulting in sclerosing lesions, with formation of pseudotumours. However, kidney involvement includes specific disease types such as TIN (IgG4‑TIN), a variety of glomerular lesions including MN, and renal pelvic lesions.3,7,10‑12Renal manifestation as the first and/ or only affectation of IgG4‑RD is rare, but some cases have been described in the literature.4
Tubulointerstitial nephritis (IgG4‑TIN) is the most common manifestation of renal involvement in IgG4‑RD presenting with declining renal function (or rapid progressive) and sub‑nephrotic proteinuria. Only few patients present with nephrotic syndrome, being membranous nephropathy the most frequent glomerular manifestation.2,3,7,11IgG4‑related MN, occurs principally in elderly male patients, extra renal manifestations of IgG4‑RD are often present, and half of the patients have concomitant TIN. Nephrotic syndrome with heavy proteinuria is the principal clinical feature in patients with IgG4‑related MN although when associated with TIN, the latter is usually the cause of renal dysfunction.10 Membranous nephropathy has been reported in patients with IgG4‑related TIN, suggesting a relation between the two autoimmune dis‑ eases, and in cases of IgG4‑related systemic disease.2,11,14
The pathogenesis of IgG4‑related MN is unclear. One hypothesis of MN developing in the setting of IgG4‑related disease is that the proliferating plasma cells produce IgG4 that is autoreactive against podocytes antigens. IgG4, does not activate complement via the classical pathway, and thus it has been proposed that the mannan‑binding lectin pathway may be involved in immune complex formation. There are alternative explanations of immune complex formation in this disease. Other IgG subclasses (particularly IgG1 and IgG3) that are co‑deposited in MN may be responsible for activating complement via the classical pathway.3,4Furthermore, type 2 helper T‑cell cytokines have been shown to cause IgG4 production by increasing and stimulating B cells in both IgG4‑related sclerosing lesions and membranous nephropathy. These aspects may explain the presence of segmental lesions of endocapillary hypercellularity as described in our case and in other types of secondary MN.10,11,15
Our patient presented with nephrotic syndrome, acute kidney injury and extreme fatigue. The presence of significant proteinuria and haematuria suggested the presence of a glomerular disease, and in addition to the IgG4‑related TIN, the kidney biopsy specimen showed concurrent membranous nephropathy with immunohistochemistry showing lymphocytes and plasma cells and IgG4 deposits in the glomerular basement membrane. The presence of IgG4‑related TIN together with the absence of circulating anti‑PLA2R antibodies support the diagnosis of MN secondary to IgG4 against coexistent primary MN. Additionally, the patient had a slightly increased IgG4 serum level with IgG and other subclasses in normal range (measured after the biopsy result). The complementary study revealed no other organ involvement to date.
The diagnosis of IgG4‑RD can be challenging and should take into consideration clinic, radiologic, serologic, and histologic aspects. To date there is no evidence of involvement of other organs other than the kidneys and typical radiological changes are not present. Therefore, renal biopsy was the key to diagnosis together with analytical changes (slight IgG4 elevation and absence of PLR2). Fulfilment of the most recent ACR/EULAR Classification Criteria for IgG4‑Related Disease (2019) supports our diagnostic hypothesis of IgG4‑related kidney disease. The patient was treated using steroids, with a decrease in proteinuria and haematuria and stabilization of kidney function.
In patients with membranous nephropathy (MN) that is accompanied by impaired renal function, elevated IgG4 levels (absolute or relative value), negative PLA2R, and/or renal interstitial plasma cell infiltration, the possibility of IgG4‑related kidney disease (IgG4‑RKD) should be carefully assessed. The good response to corticosteroid therapy favours the diagnosis of MN related to IgG4‑RD against other cause of MN.

