











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
INTRODUCTION
Multicystic dysplastic kidney (MCDK) is the most severe form of cystic renal dysplasia and is characterized by malformation of the renal parenchyma, leading to a nonreniform mass of multiple non‑communicating cysts of varying size, primitive ducts and nonrenal tissues.1‑5It is the most common cause of cystic disease in children and one of the most commonly detected anomalies on prenatal ultrasound.1,3Kidney ultrasound features include increased echogenicity, poor corticomedullary differentiation, and parenchymal cysts.2,4The causes of MCDK are not completely known. Some studies have proposed an underlying genetic predisposition.1‑7Moreover, in 7%‑14% of pregnancies with suspected MCDK, chromosomal abnormalities and syndromes are present.4 In most cases, the natural history of MCDK, if left untreated, is involution. The risk of complications, such as malignancy, hypertension, infection or chronic kidney disease (CKD) are low, with rates comparable to that of the general population, but not negligible, especially if contralateral abnormalities are present. Thus, a systematic follow‑up of all patients is recommended. Management is usually conservative, with a focus on long‑term follow‑up to identify potential complications.1,3,5,7‑10With this study we aim to evaluate the clinical course of a group of children with MCDK in a pediatric nephrology unit of a tertiary pediatric hospital between 2011 and 2023 and compare these results with previous studies conducted between 1989 and 2000 and between 2000 and 2010 at the same unit.11
METHODS
A retrospective analysis of medical records was per‑ formed, including all children with the diagnosis of MCDK (established through the identification of typical findings on ultrasound), that started follow‑up at our unit between January 2011 and December 2023. The variables included in the analysis were: gender, age at the first appointment, prenatal or postnatal diagnosis, previous follow‑up and referral, personal and family his‑ tory (with a focus on other renal abnormalities or extra‑ renal malformations), laterality of MCDK, imaging tests performed, evolution (involution of involved kidney, compensatory hypertrophy of contralateral kidney and clinical complications) and treatment. Partial involution was defined as a reduction in size of MCDK, and complete involution was defined as MCDK no longer identified in ultrasound. Compensatory hypertrophy was defined as the bipolar diameter of the contralateral kidney greater than two standard deviations of the mean value for the age. Lastly, we compared our results with two previous studies conducted in the same unit: the first between 1989 and 2000, and the second between 2000 and 2010.11 The statistical analysis was performed using the IBM SPSS Statistics® for Windows, Version 25.0 software. Values of p<0.05 were considered statistically significant.
RESULTS
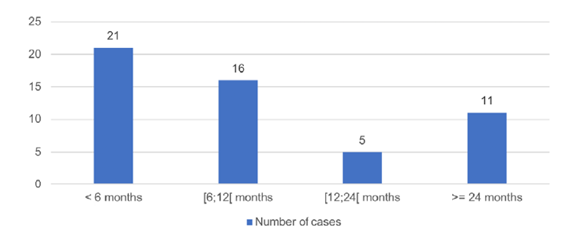
Fiftythree children were included in this study, 28 (52.8%) of which were female. The diagnosis was made during prenatal ultrasound in 48 (90.6%). The median age at the first Pediatric Nephrology appointment was six months (0‑128 months; Fig. 1), but 48 (90.6%) children were previously being monitored by other specialties, either from our hospital or from other hospitals.
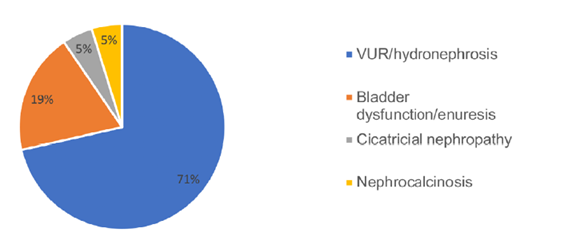
Twenty‑one cases (39.6%) had other kidney abnormalities, with the most common being vesicoureteral reflux (VUR) / hydronephrosis (Fig. 2). Extrarenal malformations and syndromes were present in eight children (15.1%), including tetralogy of Fallot, Di George syndrome, branchio‑oculo‑facial syndrome and other polymalformative syndromes. Family history of renal disease was present in 15 children (28.3%), renal agenesis being the most common (five cases), followed by nephrolithiasis, MCKD, renal duplication and pelvic kidney.
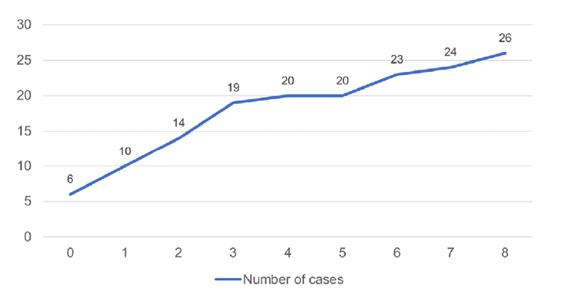
No cases of bilateral MCDK were reported and the left kidney was involved in 28 cases (52.9%). Reno‑vesical ultrasound was performed in all children. Other diagnostic tests were also performed in 50 children (94.3%): Tc‑99m DMSA scintigraphy in 29 (54.7%), 99mTc‑MAG3 scintigraphy in 28 (52.8%), and voiding cystourethrography in 28 (52.8%). Antibiotic prophylaxis was prescribed in 24 children (45.3%). Five children underwent surgery (three for the treatment of ureterocele and two because of contralateral pathology). Involution of the affected kidney was observed in 51 cases (96.2%), with a complete involution in 26 (49.1%). The median age of complete involution was two years, and the child was younger than five years in 20 cases (Fig. 3). Contralateral hypertrophy was documented in 35 cases (66.0%). Complications were reported in 18 children (34.0%): urinary tract infections in 12 cases (22.6%), proteinuria in four (7.5%), CKD in three (5.7%) and hyper‑ tension in one (1.9%). There were no reported cases of malignancy.
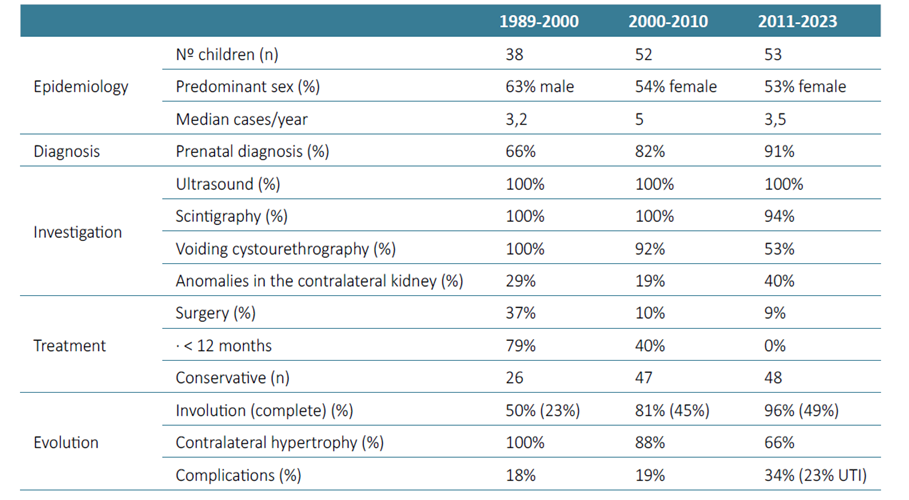
During the study period, seven children (13.2%) were discharged, four of which had reached the end of the pediatric age and were referred to adult Nephrology or to their Family Physician. Table 1 compares the results of this study with two earlier ones conducted in the same Pediatric Nephrology Unit between 1989 and 2000 and between 2000 and 2010.11
DISCUSSION
The incidence of MCDK ranges from 1:1000 to 1:4300 and previous studies showed the MCDK is more common in boys, and most cases are unilateral, more frequently involving the left kidney.1‑5,7,9In our study, there was a small female predominance (53%), and the left kidney was the most affected (53%), with no cases of bilateral disease.
The incidence of associated anomalies ranged from 5%‑48% in previous studies, including contralateral vesicoureteral reflux (the most frequent), ureteropelvic or ureterovesical junction obstruction, and less frequently, ureterocele and horseshoe kidney.3,4,9These findings are similar to those found in this study: incidence of 40%, vesicoureteral reflux being the most common.
MCDK is the most frequent renal cystic disease diagnosed antenatally, as seen in our study.1‑3,9Postnatally, it is usually diagnosed in the context of abdominal mass or by incidental identification by ultrasonography.1‑3,5Extrarenal malformations are relatively common (15% in this study) but should raise the concern for a genetic etiology. They include cardiac, central nervous system and gastrointestinal anomalies, among others.1,4
Outcome of renal dysplasia is excellent, particularly if the contralateral kidney has a normal function. The rate of malignant transformation and hypertension is comparable to that of the general population (0% and 2% in our study, respectively).1,3,8,10
The natural history is involution of the affected kidney in most cases. The rate of involution is generally greatest during the first two years of life and is completed by five years in 60% of cases.1‑4,9Our study showed a slightly lower rate of complete involution at five years (38%), but there was a higher rate of partial and complete involution compared with the previous study periods, with almost all of the children (96%) showing some degree of involution. If the contralateral kidney has no abnormalities, it usually undergoes compensatory hypertrophy, which is typically observed by three years of age.1,3,4,9,10The absence of compensatory hypertrophy suggests abnormalities of the contralateral kidney.1,4,9There was a clear decrease in the compensatory hypertrophy rate over the three study periods (from 100% to 66%). Although there is no clear explanation for this finding, this decrease may be associated withs higher rates of concomitant pathology in the contralateral kidney (40%), and complications (34%) found in the last study period. The number of children with comorbidities in the contralateral kidney and complications may have increased in our unit due to the less invasive approach adopted in recent years, which allowed children with a more benign course of the disease to maintain follow‑up with their Family Physician or attending Pediatrician, without the need for referral to a subspecialty.
Considering its favorable prognosis, algorithms for follow‑up of MCDK have shifted to less aggressive testing in the last years and affected individuals are now typically managed conservatively.1,3,5,7,8,10However, long‑term follow‑up is required, especially in patients with contra‑ lateral abnormalities, and should include serial ultrasonographic evaluation to monitor contralateral kidney growth, as well as MCDK involution, and routine follow‑up including evaluation of blood pressure, proteinuria, and kidney function studies.1‑3,5,7,8The periodicity of ultrasound and laboratory evaluations is still controversial and should be directed individually.
The same is true for the use of voiding cystourethrography or nuclear medicine renal scans.1,3,7Although contralateral VUR has been commonly reported, voiding cystourethrography is usually unnecessary in patients with normal serial ultrasound scans of the contralateral kidney, as clinically significant reflux is very unlikely in these cases. Although renal scintigraphy is usually performed in many centers, ultrasonography is generally sufficient for the diagnosis and management of MCDK, without the need for confirmation with other methods.1,2These tests should be performed, however, in cases of contralateral hydronephrosis, history of urinary tract infections or to rule out other diagnosis.1,2All children included in the three study periods underwent serial ultrasonography evaluation. Although the use of other imaging techniques was still very common, a shift towards a less invasive approach was also observed throughout the three study periods, especially regarding the use of voiding cystourethrography (from 100% to 53%).
Before antenatal ultrasound was routinely used, MCDK was most diagnosed at physical examination, as a large and palpable mass, which led to the preferred treatment of nephrectomy. In more recent years, the prevalence of MCDK has increased and the size of affected kidneys has decreased. This fact, and the knowledge of its usually good prognosis, have shifted the management to a non‑surgical approach, which was also clear in our study (rates of surgical procedures went from 37% to 9%).1,3‑5,7,10
CONCLUSION
MCDK is a severe form of cystic renal dysplasia, commonly diagnosed antenatally, but with a usually good prognosis. In recent years there was a shift towards less invasive management, emphasizing the importance of long‑term follow‑up, including serial ultrasonographic evaluation, blood pressure monitoring and kidney function studies.
TAKE HOME MESSAGES
The incidence of MCDK varies widely, and it is more frequently unilateral, with a higher prevalence in the left kidney. Associated anomalies, especially vesicoureteral reflux, are common. MCDK is typically diagnosed antenatally or postnatally through ultrasound, with extrarenal malformations occurring in 15% of cases. The prognosis for MCDK is generally excellent, with most affected kidneys undergoing involution, particularly within the first five years of life. Management of MCDK has shifted to less invasive approaches, emphasizing long‑term follow‑up with serial ultrasounds, blood pressure monitoring, and kidney function tests, reducing the need for surgical interventions.

