











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
INTRODUCTION
AA amyloidosis is a rare disease caused by tissue deposition of amyloid fibrils derived from serum amyloid A, an acute-phase reactant produced by the liver.1-3
Amyloid nephropathy is the most common presentation of AA amyloidosis, manifesting as either proteinuria or progressive kidney dysfunction. The deposition of amyloid fibrils in the kidney takes years before the first manifestations arise. The disease progresses gradually through different stages: proteinuric, nephrotic, and uremic.2,4The progression of renal AA amyloidosis to end-stage renal disease (ESRD) ranges from 27.1% to 61%.5-7
The incidence of AA amyloidosis has been decreasing, especially in developed countries, due to declining rates of chronic infections and new treatment strategies for inflammatory conditions, including autoimmune diseases. However, it continues to be a significant cause of morbidity and mortality if left untreated.8
We hereby present a case of an unusual presentation of renal AA amyloidosis, seldom described in the literature, manifesting with acute kidney injury (AKI) and sudden onset nephrotic syndrome.
CASE REPORT
The authors present the case of a 77‑year‑old man with a past medical history of chronic kidney disease (CKD) (G3aA2 stage) and several cardiovascular risk factors (arterial hypertension, type 2 diabetes mellitus, active smoking habits). He also had several cardiovascular comorbidities (paroxysmal atrial fibrillation, ischemic and valvular heart disease), chronic obstructive pulmonary disease and cystic bronchiectasis with frequent respiratory infections. His chronic medication included rivaroxaban, bisoprolol, empagliflozin/linagliptin, furosemide, perindopril/indapamide/amlodipine, budesonide, and aclidinium bromide/formoterol fumarate.
He was admitted to the emergency department with acute dyspnea, hypertension (220/110 mmHg), and peripheral edema. He was diagnosed with hypertensive pulmonary edema and started on non‑invasive ventilation, intravenous diuretics, and bronchodilators with significant clinical improvement. At hospital admission, the patient had a stable kidney function, with serum creatinine (sCr) of 1.37 mg/dL (reference range < 1.2 mg/dL) and urea of 44 mg/ dL (reference range 17‑ 49 mg/dL), sodium of 136 mmol/L (reference range 136 ‑ 145 mmol/L), potassium of 4.10 mmol/L (reference range 3.5 - 5.0 mmol/L) and a urine dipstick with protein +++ and microscopic hematuria +. His serum hemoglobin was 16.8 g/dL, white blood cell (WBC) count of 16 900 cells/µL (reference range from 4000 - 10 000/µL), he had a normal serum albumin (4.1 g/dL; reference range > 3.5 g/dL) and a low C‑reactive protein (CRP) level (0.62 mg/dL; reference range < 0.5 mg/dL). Despite an initial favorable evolution, the patient remained hypoxemic, requiring supplementary oxygen by nasal cannula and was subsequently admitted to the Internal Medicine ward.
On the first day after admission, he developed a fever accompanied by a marked increase in CRP levels to 27.7 mg/ dL. The microbiology workup was positive for Morganella morganii on blood and urine samples, and antibiotic therapy with piperacillin‑tazobactam was started for the treatment of acute pyelonephritis. Despite hemodynamic stability, fever resolution, and improvement of infectious parameters, the patient developed non‑oliguric acute kidney injury (AKI) (KDIGO stage 3) in 48 hours, with a peak sCr of 4.21 mg/dL.
Two days later, there was a worsening of peripheral edema. The laboratory workup revealed massive proteinuria, with a raised urine protein‑creatinine ratio (11 g/g; reference value < 0.2 g/g) and albumin‑creatinine ratio (8.8 g/g; reference value < 0.03 g/g), severe hypoalbuminemia (1.6 g/dL), hypercholesterolemia (total cholesterol of 256 mg/dL; reference range of < 180 mg/dL), and microscopic hematuria. He was referred to the Nephrology department for the management of nephrotic syndrome with AKI. The immunologic study was negative for rheumatoid factor, anti‑neutrophil cytoplasmic antibodies, and antinuclear and anti‑dsDNA antibodies, with normal C3 and C4 complement component values. Serum and urine protein electrophoresis, immunofixation, and free light chain ratio did not suggest monoclonal gammopathy. The anti‑phospholipase A2 receptor antibody was also negative. Serological tests for hepatitis B, hepatitis C, and HIV were negative.
Ultrasound evaluation showed kidneys with preserved dimensions, with loss of parenchymal‑central differentiation, and no hydronephrosis, measurable lithiasis, or mass lesions were identified. Fundoscopic examination of the eye showed no evidence of hypertensive or diabetic retinopathy.
A percutaneous kidney biopsy was performed. The pathology specimen included 16 glomeruli, of which two were globally sclerosed, and two were ischemic. All glomeruli showed moderate to severe nodular mesangial expansion (Fig. 1a) due to hyaline, amorphous deposits in the vascular pole and periphery stained with Congo red (Fig. 1b). These deposits were also present in the arterioles and small arteries. Some glomeruli showed fibrous, con- centric thickening of the Bowman capsule with double contours. There was also a moderate tubular injury and interstitial edema with mild lymphocyte infiltration. There was evidence of moderate chronic lesions, with 25% of glomerulosclerosis, 30% of tubular atrophy and 30% of interstitial fibrosis. The immunofluorescence study was negative. Immunohistochemical analysis for SAA (serum amyloid‑associated) protein was positive in the glomeruli and the wall of the arterioles and small arteries (Fig. 1c), confirming the diagnosis of AA amyloidosis.
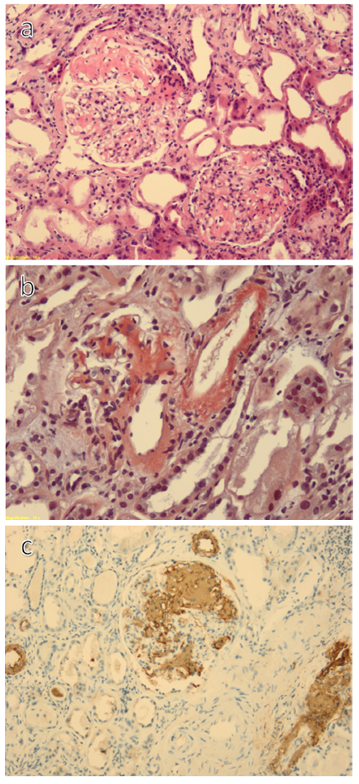
Figure 1 a) Glomerular mesangial matrix expansion with peripheral and hilar deposition of amyloid (hematoxylin&eosin, x10); b) Congo red staining, 10x; c) Amyloid A positivity in the mesangium and vascular pole, 10x;
We performed a thoracic computerized tomography that confirmed the presence of exuberant bilateral cystic and cylindrical bronchiectasis. There was no previous diagnosis or history suggestive of other disorders commonly associated with AA amyloidosis, such as chronic inflammatory arthritis or periodic fever syndrome. Echocardiography showed left atrial dilation, with a non‑dilated left ventricle, with grade 2 diastolic dysfunction and preserved ejection fraction. The patient was referred to pulmonology consultation for bronchiectasis care optimization. Serum AA levels were not available.
Early after the diagnosis of the nephrotic syndrome, supportive treatment was initiated, with a loop diuretic for hypervolemia control and angiotensin‑converting enzyme inhibitors (ACEi) for proteinuria reduction. During the hospital stay, the patient lost about 10 kg, and proteinuria decreased, with a consequent improvement of hypoalbuminemia ‑ serum albumin at discharge was 2.5 g/dL. However, the kidney dysfunction persisted with an estimated glomerular filtration rate (eGFR) of 19 mL/min/1.73 m2 and a sCr of 3.62 mg/dL at hospital discharge. Main laboratory values during patient follow‑up are reported in Table 1.
Table 1 Laboratory findings evolution during patient follow ‑up.
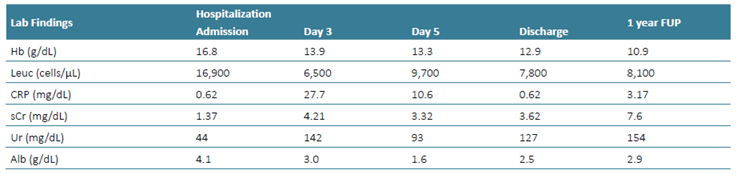
Alb - serum albumin; CRP - C ‑reactive protein; FUP - follow ‑up; Hb - hemoglobin; Leuc -leucocytes; sCr - serum creatinine; uACR - urine albumin ‑creatinine ratio; Ur - urea;
During the first year of follow‑up at our Nephrology center, there was persistence of nephrotic proteinuria and a progressive kidney function decline. Consultation on the different alternatives in kidney replacement therapy was offered. Fluid overload and symptoms of uremic syndrome, together with an eGFR of 7 mL/min/1.73 m2, led to the decision of tunneled hemodialysis (HD) catheter placement in the right jugular vein and HD initiation eleven months after the diagnosis of renal AA amyloidosis.
The HD treatments were poorly tolerated, with frequent hypotension and tachycardia episodes. Four months after the beginning of HD, the patient was again admitted to the Nephrology ward with the diagnosis of infected bronchiectasis, and two months after that was readmitted with L3‑L4 spondylodiscitis, with associated Enterococcus faecalis bacteremia. The hospitalization was complicated by intestinal ischemia, followed by hemorrhagic shock and death.
DISCUSSION
Amyloidosis is a group of diseases characterized by the deposition of insoluble beta‑sheet fibrils in extracellular tissues. This ultimately leads to organ dysfunction due to disruption of the organ architecture and direct toxicity of amyloid precursor proteins.1 Multiple amyloid proteins have already been identified as causative to amyloidosis, with immunoglobulin light chain‑derived (AL) and serum A protein being the most common.9
Renal amyloidosis is the most frequent manifestation of AA amyloidosis. The prevalence of renal amyloidosis was estimated at 2% in a retrospective study that reviewed native kidney biopsies from 474 patients of the Mayo Clinic.10 AL amyloidosis was the most frequent type of amyloidosis found in the Mayo Clinic and in a German registry, followed by AA amyloidosis.10,11There is, however, significant geographic variability in the distribution of the various types of amyloidosis: a Portuguese retrospective cohort study that identified patients with renal biopsy‑proven amyloidosis found that AA amyloidosis was the most common form (56.1%) and that chronic infection was the leading cause of the disease in almost half of the cases.12
The clinical manifestations of renal amyloidosis are wide. Albuminuric proteinuria is the most frequent presentation and can vary from the sub nephrotic range to massive proteinuria that results in nephrotic syndrome - the latter is more typical of AL amyloidosis.10 In patients with AA amyloidosis, the predominant disease manifestation is slowly progressive chronic kidney disease.
In AA amyloidosis, the protein responsible for the pathogenic disturbance in kidney architecture and function is SAA protein, an acute-phase reactant produced by the liver. The increase in serum concentration of SAA protein due to elevated levels of cytokines in chronic inflammatory states, together with the conformational changes in the protein, leads to aggregation and kidney deposition.1The most frequent underlying disorders of AA amyloidosis are chronic inflammatory arthritis, chronic sepsis (of which bronchiectasis is one variant), and periodic fever syndromes, such as familial Mediterranean fever (FMF).
We presented a case of a patient with renal AA amyloidosis. After the diagnostic workup, we presumed chronic bronchiectasis to be AA amyloidosis’ most probable underlying cause. The clinical presentation here depicted is, however, atypical. Instead of progressive kidney dysfunction, the presentation was characterized by AKI and a sudden‑onset nephrotic syndrome.
The concept of “amyloid storm” was applied to describe a rare clinical manifestation of AA amyloidosis manifestation in a group of FMF patients, followed at an Israeli center.4 The criteria employed by this group for diagnosing “amyloid storm” in FMF patients were a sudden increase in urinary protein excretion and serum creatinine in less than 2 weeks, with a simultaneous rise in CRP to levels at least 10 times the highest normal value. All these criteria were fulfilled by the patient described in this case.
The pathophysiology of “amyloid storm” is unknown. Still, an infection not necessarily related to the chronic sepsis underlying AA amyloidosis may be responsible for the sudden overproduction and deposition of amyloid - pro‑inflammatory cytokines released during acute infection responses may upregulate AA amyloid production. Macrophages are responsible for the cleavage of plasma high‑density lipoproteins, where SAA circulates,13 possibly also contributing to the release of higher concentrations of this protein during infection. The direct toxicity of amyloid precursor proteins and folding intermediates also contribute to the clinical findings and can explain the disease manifestations even without extensive amyloid deposits.1,14A Turkish retrospective study evaluated the histopathological findings associated with rapidly progressive renal failure in AA amyloidosis: global amyloid deposition, vascular pole involvement, peritubular amyloid deposition, and severe interstitial inflammation were identified as risk factors for the need for renal replacement therapy one year after diagnosis.15 Some of these histologic characteristics were also present in the renal biopsy performed in our patient.
The treatment for AA amyloidosis is primarily directed to the underlying cause of AA production, as managing the underlying inflammatory conditions can reduce or reverse the disease.16 Antibiotics, surgery, and respiratory physiotherapy are commonly used to reduce the disease burden in chronic bronchiectasis, but with variable outcomes.17 The cytokines IL‑1, IL‑6, and TNFa mediate the production of SAA18 and are recognized pharmacologic targets. Multiple anti‑cytokine agents are currently being tested for the treatment of renal AA amyloidosis, even when the cause of SAA production is unknown.8,19Nevertheless, immunosuppressive side effects of such therapies may contribute to aggravation of chronic inflammatory background through increased infectious risk. At this date, there is no compelling evidence of the benefit of routine use of anti‑cytokine agents for AA amyloidosis.
Survival on dialysis is poor in patients with AA amyloidosis, with a reported median survival of 20 months.20 Cardiac and autonomic dysfunction usually present in multisystemic amyloidosis contribute to difficulty in the management of these patients with hemodynamic fragility.1 This autonomic dysregulation then limits the use of diuretics or ultrafiltration. When the presentation of renal amyloidosis occurs in the form of an “amyloid storm”, the prognosis is poorer, with 80% of the patients reaching ESRD or death one year after the diagnosis.4
CONCLUSION
Renal AA amyloidosis is a rare disease with multiple presentations at diagnosis, one of which is the scarcely described “amyloid storm”. We report one of the few cases described in the literature of this entity presenting with AKI and nephrotic syndrome. This atypical form of the disease has a rapid progression and is associated with poor renal and vital prognosis. However, more studies on the pathogenesis of this disease are needed to guide the treatment and implement strategies to delay its progression and improve outcomes.

