











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
INTRODUCTION
Autosomal dominant tubulointerstitial kidney disease (ADTKD) is an underrecognized condition that accounts for approximately 2%‑5% of monogenic causes of chronic kidney disease (CKD).1 It is characterized by tubular damage and interstitial fibrosis, typically in the absence of significant glomerular involvement.2 Disease‑causing mutations have been identified in four genes encoding uromodulin (UMOD), renin (REN), mucin-1 (MUC) and hepatocyte nuclear factor-1B (HNF1B).3 More recently, mutations in α1 subunit of the SEC61 translocon (SEC61A1) and DNAJ/ HSP40 homolog, subfamily B, member 11 (DNAJB11) have also been implicated in ADTKD, further expanding the genetic spectrum of the disease.4HNF1B is a transcription factor essential for the embryonic development of kidneys, liver, pancreas, and urogenital tract. Mutations in HNF1B gene can lead to a multisystemic disorder encompassing CKD, congenital anomalies of the kidney and urinary tract (CAKUT), maturity-onset diabetes of the young (MODY5), hypomagnesemia and hyperuricemia.5,6Here, we present the case of a pregnant woman with CKD, renal cysts, electrolyte abnormalities, a uterine malformation, and a positive family history of CKD, in whom genetic testing revealed a novel heterozygous mutation in HNF1B.
CASE REPORT
A 37‑year‑old pregnant woman at 26 weeks of gestation, with stable CKD stage G3a (estimated glomerular filtration rate of 54 mL/min/1,73 m2) previously attributed to chronic pyelonephritis, presented to the emergency department with acute pyelonephritis. Her medical history was significant for gestational diabetes, hyperuricemia disproportionate to her CKD stage, and two prior urinary tract infections. Notably, her mother had been diagnosed with CKD of unknown etiology and had been on hemodialysis since the age of 50. No other family members were known to have CKD (Fig. 1). On presentation, laboratory findings included a serum creatinine of 1.23 mg/ dL, normal liver function tests, hyperuricemia of 7.3 mg/ dL (despite treatment with 100 mg allopurinol), hypomagnesemia of 0.93 mEq/L, and renal tubular acidosis with serum bicarbonate of 18.5 mmol/L. Urinalysis revealed an unremarkable urine sediment and absence of proteinuria. A renal ultrasound demonstrated bilateral renal dysplasia, with normal‑sized kidneys displaying increased echogenicity and multiple bilateral cortical cysts. An abdominal ultrasound identified a septate uterus.
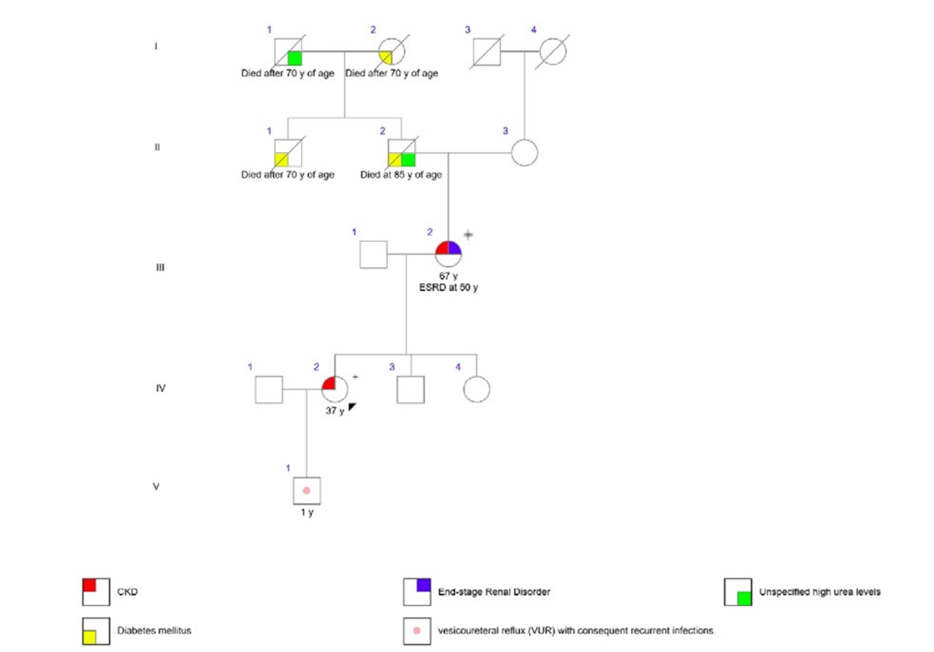
Figure 1. Pedigree of the family, showing the main clinical characteristics. No family members besides the mother’s proband (III-2) had end-stage renal disorder (ESRD) and none died at a young age. The + sign refers to the presence of the heterozygous HNF1B c.785_786dup p.(Ala263Argfs*3) variant in DNA obtained from leucocytes in the peripheral blood.
Given these findings, HNF1B‑related nephropathy with predominant features of ADTKD (ADTKD‑HNF1B) was suspected. Genetic testing was performed on leukocytes obtained from peripheral blood using a targeted next‑generation sequencing (NGS) panel on the Illumina NovaSeq6000 platform (Macrogen). The panel included the genes HNF1B, MUC1, REN, UMOD, COL4A3, COL4A4, COL4A5, SEC61A1, and SLC8A1. This analysis identified a previously undescribed heterozygous variant in HNF1B, c.785_786dup p.(Ala263Argfs*3), classified as likely pathogenic according to the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) criteria.7 Subsequent genetic testing of the patient’s mother identified the same mutation, supporting the diagnosis of ADTKD‑HNF1B in this family. Despite the episode of pyelonephritis and a diagnosis of gestational diabetes, the remainder of the pregnancy was uneventful. The patient underwent spontaneous labor at 37 weeks and 1 day, following spontaneous rupture of membranes, and delivered a healthy male infant. During the first months of life, the newborn experienced urinary tract infections due to vesicoureteral reflux; however, genetic testing for the familial HNF1B mutation was negative.
DISCUSSION
We present a case of a young woman with CKD previously attributed to chronic pyelonephritis, who was diagnosed during pregnancy with ADTKD‑HNF1B due to a novel heterozygous variant c.785_786dup p.(Ala263Argfs*3) at HNF1B gene. Subsequent familial screening revealed that the patient’s mother, who was on hemodialysis for CKD of unknown etiology, carried the same mutation, leading to the recognition of familial ADTKD‑HNF1B. Mutations at HNF1B can result in multisystemic manifestations, including renal, hepatic, neurological, pancreatic and genital anomalies. As a key transcription factor in nephron development, HNF1B variants are a recognized monogenic cause of CAKUT, often presenting with bilateral kidney cysts, resembling autosomal dominant polycystic kidney disease. In addition to CAKUT, patients may present with isolated hypomagnesemia, ADTKD, or extra‑renal manifestations, making HNF1B-related disease a preferred term.5,6,8This phenotypic variability that cannot be predicted by the genotype, aided by its variable penetrance, high intra‑/interfamilial heterogeneity, contributes to diagnostic challenges and may lead to an underestimation of disease prevalence.9 Moreover, molecular diagnosis is further complicated by a high frequency of de novo mutations (>50%), which can discourage genetic testing, and copy number variations, which may not be detected using next‑generation sequencing techniques.10 Despite not being previously described, the c.785_786dup (Ala263Argfs*3) frameshift mutation identified in this case results in a premature termination codon, likely leading to the absence or truncation of the HNF1B protein, which explains the patient’s phenotype. This supports its classification as a likely pathogenic variant.7 Our patient was referred for genetic counselling, given the 50% risk of transmission in future pregnancies. She continues to receive CKD management following established guide-lines, with special attention to diabetes and electrolyte imbalances. If kidney transplantation is considered in the future, disease recurrence in the allograft is not expected. However, related living donors should undergo genetic testing to assess for potential disease transmission. In conclusion, this case underscores the clinical spectrum of ADTKD-HNF1B and highlights the importance of genetic testing in diagnosing monogenic kidney diseases. By identifying pathogenic variants, genetic testing enhances disease awareness among nephrologists, facilitates familial screening, and enables genetic counselling, ultimately improving patient care.

