











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
INTRODUCTION
Systemic sclerosis (SSc) is a rare autoimmune disease characterized by immune system dysfunction, associated with the production of autoantibodies and cell‑mediated autoimmunity. It compromises the connective tissue of the skin and involves endothelial dysfunction and fibroproliferative vasculopathy, leading to the involvement of vital organs.1 The disease has a complex etiology, likely related to both environmental and genetic factors. It is well‑established that immune and vascular processes play a central role in the pathogenesis of SSc.2 However, it remains unclear what the initial events are that trigger the fibrotic process and vasculopathy seen in the disease.3 The condition has an estimated annual incidence of 8 to 56 new cases per million and a prevalence of 38 to 341 per million, based on evaluations of the adult population.4 It predominantly affects women, with 3.8 to 15 times more cases in this gender.5 Despite improvements in the recognition and treatment of organic involvement, SSc remains the collagenosis with the highest mortality rate.6 It can affect the skin, interstitium, pulmonary arteries, gastrointestinal tract, heart, and kidneys.1,6In this study, we report a patient who, after being treated with cyclophosphamide at high dose over the preceding two months for interstitial pulmonary involvement in systemic sclerosis, developed severe renal dysfunction requiring renal replacement therapy due to the onset of ANCA-associated vasculitis (AAV). The unusual onset and progression of this dysfunction required a thorough clinical, radiological, and histopathological assessment, leading to an adjustment in the immunosuppressive regimen, a partial response, and continued monitoring.
CASE REPORT
A 42‑year‑old woman, was transferred from the emergency department to a referral hospital with symptoms including low back pain, nausea, and subacute fever. The patient also presented with acute kidney injury, classified as KDIGO III. Four months before admission, the patient had been diagnosed with SSc, based on sclerodactyly, sporadic fever, Raynaud’s phenomenon, an anti‑SCL‑70 level of 240 (reference < 15), and pulmonary involvement manifesting as a nonspecific interstitial pneumonia, Fig. 1, extending to both the bases and middle thirds of the lungs.
Given the involvement of a vital organ, the patient was 0started on immunosuppressive therapy with the alkylating agent, cyclophosphamide (CTX), at a dose of 0.8 g/m², corresponding to a total of 1.3 g, administered monthly, with a planned duration of six months. The patient also re- ported symptoms of dyspepsia, heartburn, and occasional conduction dysphagia. Additionally, paresthesias in the lower limbs, particularly in the heel region, were noted during driving. Following the administration of the initial dose and a concomitant five‑day course of levofloxacin, the patient showed partial improvement in sweating, evening chills, arthralgia, and cough. The schedule for the second dose of CFX was maintained. However, after the therapy, the patient experienced a decline in overall condition, accompanied by low back pain, recurrence of fever, nausea, vomiting, and loss of appetite, prompting a medical consultation. Acute KDIGO III kidney injury was confirmed, along with suspected pyelonephritis and scleroderma renal crisis. Ceftriaxone 2 g/day and captopril 12.5 mg every 12 hours were initiated, and the patient was transferred to a tertiary hospital. During this period, the patient had three negative sputum smear microscopies.
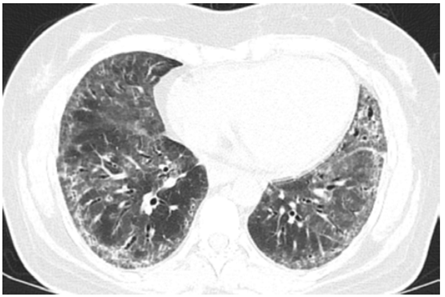
Figure 1 Axial section of chest computed tomography (CT), revealing thickenning of bronchovascular bundles, reticular opacities and difuse bilateral ground‑glass opacities.
Upon admission to a referral hospital, the patient had a creatinine level of 6.15 mg/dL, urea of 72 mg/dL, protein-uria of 1208 mg/24h, microscopic hematuria with erythrocyte dysmorphism, and red blood cell casts. Empiric antibiotic therapy, which had been started in the emergency room, was continued, and admission tests were ordered. An angiotensin‑converting enzyme (ACE) inhibitor was discontinued due to the worsening of renal function. By the fifth day, the patient developed symptoms of uremia, necessitating the initiation of renal replacement therapy. Given the progression of the case, which was initially refractory to ACE inhibitors and lacked thrombocytopenia, hemolysis and the difficult‑to‑control hypertension typical of 90% of renal crisis cases in scleroderma, a renal biopsy was performed. Blood and urine cultures were negative. On the eighth day of hospitalization, the patient received the third dose of cyclophosphamide, continuing the immunosuppressive regimen. The patient developed symptoms such as chills and a subfebrile temperature, indicating a decline in overall health. Laboratory tests revealed severe neutropenia and abdominal tomography with gas foci observed in a biopsied kidney, suggesting the presence of a perirenal hematoma or renal abscess. Consequently, antibiotic therapy with teicoplanin and meropenem was initiated, and the initial methylprednisolone regimen was suspended. A subsequent urine culture revealed the presence of Klebsiella pneumoniae, with procalcitonin levels measuring 0.52 ng/mL (reference range: up to 0.1 ng/mL). Additionally, there was a gradual improvement in neutropenia, accompanied by intermittent fever, as indicated by an increase in the leukocyte count. A pathological analysis of the renal biopsy specimen (Fig. 2), revealed the presence of glomerular crescents, affecting more than 50% of the evaluated glomeruli. A significant limitation was the representativeness of the renal tissue sample, which included both cortex and medulla but was limited to two glomeruli and lacked vascular structures, thereby precluding the anatomopathological exclusion of a scleroderma renal crisis. These crescents were accompanied by early interstitial tubule fibrosis, which was associated with a pauci‑immune pattern on immunofluorescence. Antineutrophil cytoplasmic anti-body (ANCA) testing by indirect immunofluorescence was positive for the pANCA pattern, with a titer of 1:320 (reference value: non‑reactive). The patient’s sample was further tested and found to be positive for antibodies against myeloperoxidase, with a titer of 80 IU/mL.
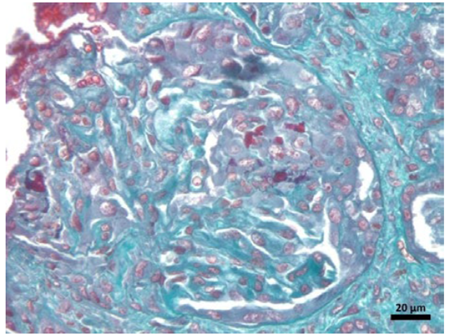
Figure 2. Tissue sample with Masson’s trichrome stained edge under optical microscopy. The renal anatomopathological findings included glomeruli with progressive necrosis, as well as tubulointerstitial fibrosis and moderate tubular atrophy. No vessels in the sample are available.
New cultures showed no bacterial growth, and the absence of signs of catheter-associated bloodstream infection justified the discontinuation of antimicrobials. After 48 hours of clinical stability, pulse therapy was initiated with methylprednisolone 1 g/day for three days, followed by rituximab at a dose of 375 mg/m² weekly for four weeks, considering the patient’s condition of ANCA vasculitis with severe renal involvement. The patient continued with prednisone at 1 mg/kg/day after methylprednisolone, and one week after the fourth rituximab infusion, at a dose of 50 mg/day was initiated to facilitate corticosteroid tapering, considering the associated risks of corticosteroid therapy in patients with scleroderma. The patient’s condition remained stable, with an increase in diuresis. After seven days of pulse therapy with methylprednisolone, the frequency of renal replacement therapy (RRT) sessions was reduced to twice per week, with consistent control of volume, electrolyte balance, and acid‑base parameters. By the end of the fourth week of rituximab treatment, the frequency of hemodialysis was further reduced to once a week. Subsequent follow‑up visits revealed that the hemodialysis catheter could be removed five days after hospital discharge, with an estimated glomerular filtration rate of approximately 21 mL/ min/1.73 m². This indicated a partial response of crescentic glomerulonephritis due to ANCA vasculitis associated with systemic sclerosis. The patient gave consent for the presentation of the case and the data included.
DISCUSSION
Systemic sclerosis (SSc) is a multisystem disease, with renal involvement being particularly significant due to its high morbidity and mortality. While renal scleroderma crisis (SRC) is the most well‑known form of renal impairment associated with SSc, affecting approximately 10% of patients with the disease,9 it is important to consider other differential diagnoses, including anti‑neutrophil cytoplasmic antibody (ANCA)‑associated vasculitis, in this patient population. In 2020, a patient diagnosed with SSc four years earlier presented with kidney dysfunction, nephrotic syndrome, and hypertension, mimicking SRC. However, renal biopsy, ANCA and anti‑MPO titers, along with a partial response to corticosteroid therapy, supported the diagnosis of vasculitic renal involvement.10Renal scleroderma crisis typically manifests within the first four years of diagnosis in patients with systemic sclerosis, while ANCA vasculitis usually presents later, on average, after seven years.11 In the case presented here, the patient had been diagnosed four months prior, with immunosuppression initiated two months before the renal dysfunction. The patient exhibited normotension, active urine sediment, urine protein above 1 g/24h and biopsy findings showing glomerular involvement in a pauci‑immune manner, leading to the diagnosis of an overlap syndrome in this patient, who had been diagnosed with scleroderma less than one year earlier. The prevalence of ANCA‑associated vasculitis in the scleroderma population is estimated to be rare, with a range of 2.5% to 9%, particularly in patients with the limited cutaneous form.10 The prevalence of connective tissue diseases superimposed on vasculitis ranges from 11% to 12%, with vasculitis limited to the kidneys and rheumatoid arthritis being the most prevalent. In Syria, a similar case has been reported, where a patient with systemic sclerosis (SSc) and severe renal dysfunction was being treated for pulmonary hypertension and interstitial pulmonary disease.12,13Notably, this patient exhibited the presence of ANCA and anti‑MPO, although no correlative anatomopathological analysis was available. A distinguishing feature of this case was the lower intensity immunosuppression administered.
Some studies have reported cases of patients with renal involvement in the form of ANCA-associated glomerulonephritis, presenting similarly and developing severe complications despite being immunosuppression‑naive.14 However, in our case, the patient was still undergoing induction therapy with high‑dose cyclophosphamide-a drug with a well‑established role in this context-which further contributed to the atypical nature of the presentation, hindering early recognition of the condition and delaying adjustments to the immunosuppressive regimen. A report from Iran describes a patient with systemic sclerosis who received immunosuppressive treatment and subsequently developed renal involvement.15 In contrast to our patient, the Iranian patient’s immunosuppressive regimen was maintained with low doses of corticosteroids and azathioprine. The renal involvement in the Iranian patient was nephrotic syndrome, accompanied by focal segmental glomerulosclerosis, which was not associated with ANCA.
One of the most significant challenges in managing this case was distinguishing autoimmune inflammatory activity from infectious complications, which led to a delay in the initiation of adequate immunosuppression. The presence of fever, elevated C‑reactive protein (CRP), and procalcitonin raised the possibility of infectious complications, particularly given the patient’s history of immuno- suppression. The delayed diagnosis of systemic sclerosis and initiation of immunosuppressive therapy, along with the prolonged turnaround time for diagnostic markers such as ANCA and anti‑MPO, underscore limitations that are, at times, inherent to the Brazilian public healthcare system. The switch from cyclophosphamide (CFX) to rituximab was based on the observation that, at the time CFX was initiated for interstitial lung disease, the patient exhibited normal urinary function and sediment; the onset and progression of the renal condition occurred during more than three months of CFX therapy. Although avacopan was considered, access to this medication is highly restricted in Brazil, particularly in the Northeast region, being limited to research protocols or legal proceedings. The corticosteroid dose was selected based on treatment protocols for similar cases of granulomatosis with polyangiitis with renal involvement, typically ranging from 500 to 1000 mg per dose.
It is worth mentioning that several clinical features can suggest a diagnosis of AAV rather than scleroderma renal crisis (normal or slightly higher blood pressure, red blood cell cast, urine protein > 1 g/day, normal platelet measurement), but a renal biopsy without signs of arterial proliferative vasculopathy or thrombosis is important for accurate diagnosis. In addition, the importance of rituximab as a therapy to induce and maintain remission in cases of overlap syndrome between systemic sclerosis and associated ANCA vasculitis is highlighted, particularly in cases where there is a profile of adverse effects related to cyclophosphamide use or failure of remission therapy. Further studies are needed to evaluate, in the long term, the ability of alternative therapies to cyclophosphamide to sustain remission in such autoimmune diseases.

