











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
INTRODUCTION
Atypical hemolytic uremic syndrome (aHUS) is a complement‑mediated thrombotic microangiopathy caused by genetic mutations or antibodies against complement factors, leading to uncontrolled activation of the alternative pathway of the complement system.1-3
Loss of function mutations of complement factor H (CFH) are the most common genetic cause of aHUS; however, other non-complement-related pathogenic variants have also been identified as causes and are not yet routinely screened for. In cohorts tested for such, the incidence is 2%‑3%4,5and their detection can change the course of treatment.6
In the Global Registry of aHUS, approximately 40% of the 851 patients had no mutations identified.1
Several studies have elucidated the possible correlation between genotype and phenotype in renal manifestations.4 The most benign mutation is the one occurring in membrane cofactor protein (MCP), leading to end-stage renal disease (ESRD) in 5 years in 10%‑50% of cases, as opposed to mutations in CFH, leading to ESRD in 70%‑80% of cases.4,5
Before eculizumab approval in 2011, supportive treatment with plasma exchange (PE) and fresh frozen plasma reposition rendered a high probability of death and ESRD.1,7,8Early target treatment with eculizumab has reduced the overall mortality rate below 5%, and increased renal recovery rates above 70%.8,9
In this clinical case, we report a patient diagnosed with aHUS and treated with eculizumab. Six months after remission, eculizumab was successfully withdrawn. We aim to discuss the need for an individualized treatment strategy, considering etiopathogenesis and the morbidity and costs associated with lifelong treatment with eculizumab.
CASE REPORT
We report a clinical case of a 57-year-old Caucasian woman with a medical history of pharmacologically controlled long-term hypertension, type 2 diabetes mellitus with no target organ lesions described, dyslipidemia, and depressive syndrome. Chronic medications included an association of losartan plus hydrochlorothiazide, atorvastatin, paroxetine, and an association of metformin plus canagliflozin.
She had had a healthy pregnancy and declared no alcohol or smoking habits. Her oncologic screenings were relevant for a negative uterine cytology and a negative fecal occult blood test - test-immunochemical test. Her previous plasmatic creatinine, 4 months earlier, was within normal range (0.58 mg/dL).
She sought the emergency service because of watery diarrhea, vomiting, and worsening asthenia, which had been ongoing for 5 days. She denied other symptoms, namely fever, respiratory or urinary symptoms, or arthralgia. She did not report other sick cohabitants and denied recent traveling, drinking/eating from possible contaminated sources, and taking over the counter medications. Physical examination was only relevant for mild hypotension. No cutaneous alterations were visible.
In Tables 1 and 2 , we present the most relevant results obtained during patient management.
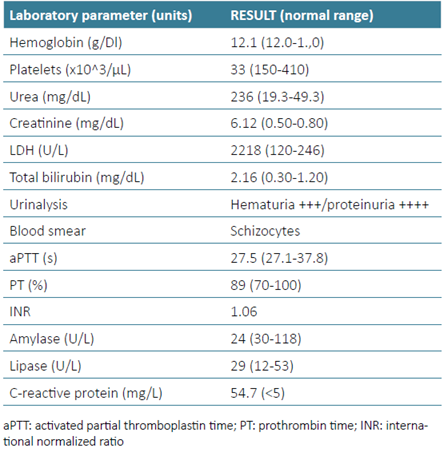
Laboratory investigation was conducted to better understand her clinical status, and it indicated a severely low platelet count, very high levels of LDH, and a discrete elevation in bilirubin levels. Peripheral blood smear revealed the presence of rare dacrocytes and schistocytes, with no platelet aggregates. C‑reactive protein was modestly increased. A marked elevation of serum creatinine and blood urea nitrogen (BUN) was noted, corresponding to an Acute Kidney Injury Network (AKIN) classification grade 3, along with hemoglobinuria and low‑grade proteinuria. A renal ultrasound excluded obstructive causes of acute kidney injury (AKI) and echographic signs of chronic kidney disease.
Discarding infection, blood cultures, and urine cultures were promptly collected. The rapid test for the detection of Escherichia coli O157 in feces was negative.
She was kept under observation, and in the following 24 hours, her clinical condition worsened, evolving with oliguria, hypertension, and mental confusion.
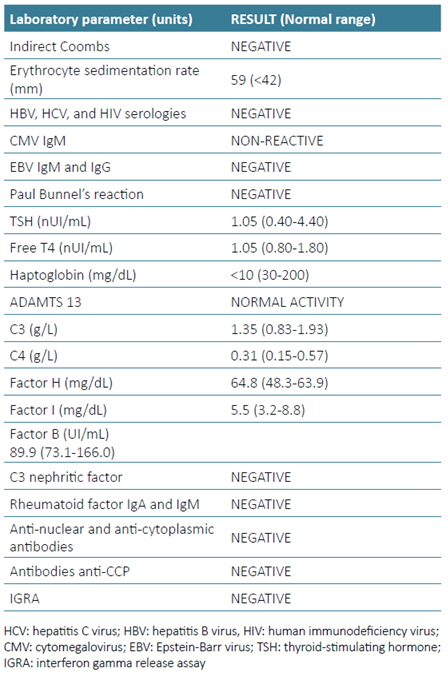
Analytic reevaluation showed an aggravating serum creatinine and BUN, and a more severe thrombocytopenia. Anemia was evident, with a hemoglobin level of 10.2 g/dL, and LDH levels continuously increasing. Direct Coombs test and irregular antibodies (ENZ) were negative. Viral serologies for B and C hepatitis, HIV, CMV, and EBV were requested, and the results later obtained were all negative. The IGRA test excluded latent tuberculosis.
This clinical presentation and rapid evolution were consistent with thrombotic microangiopathy (TMA). Complement studies, haptoglobin levels, ADAMTS 13 activity, and autoimmune panel were later obtained. As these tests take a relatively long time to be ready, and in the face of clinical deterioration, a presumptive diagnosis of complement‑mediated TMA was established, and low‑flux hemodialysis, plasma exchange therapy, and eculizumab were started in the first 48h of presentation.
Plasma exchange therapy was performed, with 1.5 per plasma volume [60-75 mL/kg] per session, replaced with a combination of 50% human albumin and 50% fresh frozen plasma. Antibiotic prophylaxis was instituted, and she was vaccinated for Haemophilus influenzae, Streptococcus pneumoniae, and meningococcal disease ACWY 135 and B. Eculizumab was administered in a full dosage (900 mg, in a slow infusion rate). She completed a total of three daily PE sessions, accompanied by supplementary administration of eculizumab after each one (600 mg in the first and 300 mg in the subsequent sessions, respectively).
Rapid improvement was noted. Anemia had resolved, platelet count was normalized, and a trending downgrade of LDH levels was evident. She showed rapid improvement in her general condition.
Histological analysis of renal biopsy showed signs of acute and chronic TMA, namely extensive endotheliosis and mucoid edema of the intima and glomeruli with intracapillary thrombi (Fig. 1).
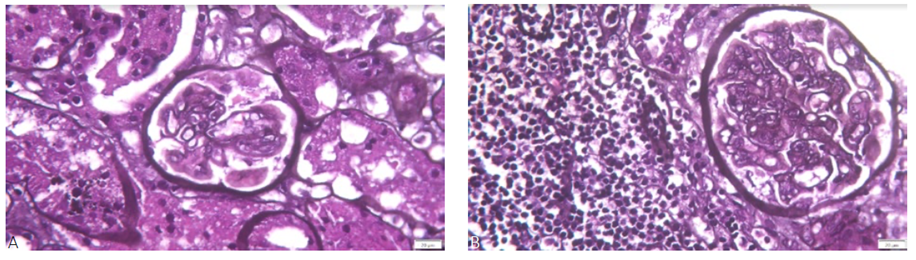
Figure 1 A. Light microscopy PAS staining showing glomerular endotheliosis (200x) and B: Ligh microscopy PAS staining showing membrane double contours (200x)
Despite excellent hematological response, the patient remained dialysis‑dependent at the time of hospital discharge for about one month. Eculizumab was maintained, weekly for 4 weeks, for the induction phase, and then in the maintenance phase, 900 mg was administered every two weeks.
Posterior results showed C3 and C4 within normal levels, as well as complement proteins factor H, factor I, factor B, and C3 nephritic factor. A mildly elevated factor H was detected. Autoimmune panel (anti‑nuclear, anti‑cytoplasmatic antibodies, and anti‑CCP antibodies) was negative. Haptoglobin was untraceable, less than 10 mg/dL, and ADAMTS 13 activity was more than 10%. These results were more in favor of a diagnosis of aHUS, confirming the initial suspicion.
A genetic test was ordered. NGS genomic sequencing test results, based on 14 genes, whole exome sequencing (WES) including copy number variation (CNV) analysis (ADAMTS13, C3, CD46, CFB, CFH, CFHR1, CFHR3, CFHR4, CFHR5, CFI, DGKE, LMNA, MMACHC, THBD) were negative for pathogenic variants.
After 6 months of monthly follow‑up and eculizumab administration, she maintained clinical remission based on hematologic and renal criteria, with normal hemoglobin, platelet count, normalized LDH, and normal serum creati-nine. No adverse effects were ever reported.
Her notable improvement and the lack of genetic mutations detected led us to consider eculizumab withdrawal. It was suspended after 6 months of treatment, and she maintained close monitoring with trimestral evaluations.
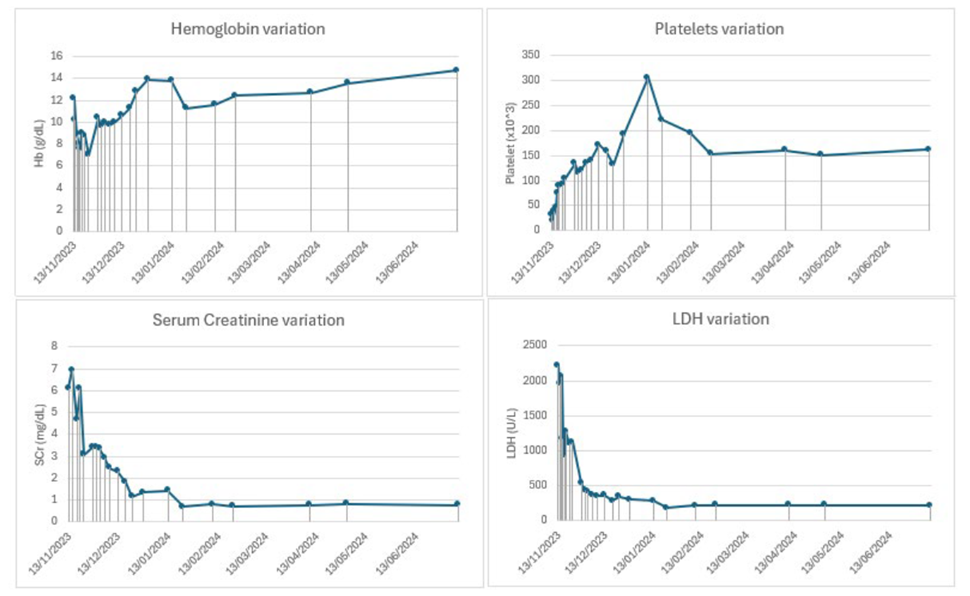
After 12 months of eculizumab withdrawal, the patient maintains hematologic and renal remission. Fig. 2 shows hemoglobin, platelets, serum creatinine, and LDH variations since admission to the last follow‑up appointment.
DISCUSSION
We report a case of a middle‑aged Caucasian woman, with a classical presentation of aHUS, triggered by a previous gastrointestinal infection, which is common in 20%‑30% of patients.3,8Secondary causes of TMA, typical HUS, and thrombotic thrombocytopenic purpura (TTP) were excluded.1 Prompt support treatment and eculizumab were initiated, leading to a clinical and analytical improvement. Eculizumab was withdrawn after 6 months of clinical remission, based on the lack of pathogenic variants in the genetic test.
Evidence for the use of eculizumab in aHUS comes mainly from two prospective phase 2 trials, with a cohort of aHUS patients older than 12 years, who initially received eculizumab for 26 weeks, and subsequently, prolonged to a long‑term extension phase, during 64 and 62 weeks, respectively. Trial 1 studied 17 patients with both low platelet count and renal damage, and the primary endpoint was the change in platelet count from baseline to week 26. Trial 2 studied 20 patients with renal damage but without a sustained decrease in platelet count, despite plasma exchange, and the primary endpoint was “TMA event‑free status,” defined as no decrease in platelet count >25%, no plasma exchange or infusion, and no initiation of dialysis. In Trial 1, there was a statistically significant increase in platelet count, and in Trial 2, 80% of patients achieved the TMA event-free status. Overall, eculizumab led to signif-icant, continuous, and time‑dependent improvement in the estimated glomerular filtration rate (eGFR).10
In terms of safety profile, treatment was not associated with major adverse events, with results being like the ones involving cohorts of patients with paroxysmal nocturnal hemoglobinuria who received treatment for up to 8 years.11
Despite that, one must not ignore the infectious risk and other adverse events. Data from the United States showed that between 2008 and 2016, 16 cases of meningococcal disease were identified in patients treated with eculizumab, and 14 of them were documented to have received at least one dose of meningococcal vaccine before the onset of disease.12
A meta-analysis conducted in 2025, encompassing nine RCTs, in a total of 691 patients, concluded that eculizumab did not significantly increase the overall risk of infection when compared to the control (placebo or standard of care), but subgroup analysis revealed that eculizumab raised the risk of urinary tract infections and severe bacteremia.13,14
Economic burden is something that must be considered, as long‑term/life‑long eculizumab administration tends to be resource-consuming consuming.8,15
Orozco-Leal et al elaborated a Markov model to estimate cost and quality‑adjusted life years (QALYs), in a scenario of eculizumab withdrawal and disease monitoring strategy. They stressed an increased average patient QALYs and reduced costs, with a low impact on survival estimates (average reduction of 0.0005 patient life years) over an 80‑year time consideration. They conclude that treatment withdrawal and disease monitoring were cost‑effective compared with lifelong eculizumab therapy, substantially reducing costs per patient and improving patient quality of life.15
Although the initial studies advised a prolonged course of treatment, data gathered provided us with reasonable arguments to adopt an individualized therapeutic plan. The most credible evidence that supports our case management comes from 9 reports, which included 171 patients with aHUS who stopped eculizumab after a median treatment of 6 months. A 12‑month follow‑up showed an overall relapse rate of 27%, with a median time of 3 months.16In those cases, treatment was promptly restarted, preventing chronic sequelae.16
A prospective multicentre study developed by Fakhouri et al, involving 55 aHUS patients with detected genetic variants, showed that the risk of relapsing after eculizumab withdrawal was mainly determined by the presence of some specific variants in complement genes, and that discontinuation is safe in patients with no such variants detected, with a relapse rate inferior to 5% in this subgroup.8 Some studies have emerged suggesting that withdrawal of eculizumab in patients with native kidneys can be successfully made after 3 months of treatment, even in the presence of a pathogenic variant or antibodies against H factor, as evidenced by the CUREiHUS study.17 In a cohort of 21 aHUS patients, 17 had a complement genetic variant or antibodies against factor H identified, and all of them were treated with eculizumab for a median duration of 13.6 weeks. Only 4 relapses occurred.17
Despite these results, the low sample size and event rate did not allow for establishing which were the predictors of relapse, underlining that large data registries analysis is needed to determine outcomes of eculizumab treatment discontinuation.17
In our case, after eculizumab withdrawal, monitoring was performed through regular assessment of hemogram, LDH, and, more importantly, urinalysis, as hemoglobinuria is the most precocious indicator of a relapse. Data from a retrospective study by Brambilia et al, including 84 patients, determined that detection of hemoglobinuria in relapsing disease showed a sensitivity of 100% and a specificity of 87.4%, with a positive predictive value of 10.5% and a negative predictive value of 100%, with an accuracy of 87.6%.18
The patient is now in complete remission, and no relapses were detected, proving in this case, a successful approach.
CONCLUSION
aHUS is a rare, life-threatening TMA driven by dysregulation of the complement system.1-3Early recognition is critical. Delays remain a major hurdle due to overlapping clinical presentations and the need for genetic and complement testing.1-3
Eculizumab revolutionized treatment by significantly reducing mortality, preserving kidney function, and lowering the risk of systemic complications. Maintaining lifelong treatment carries a high cost, lifelong susceptibility to serious infections, and possibly other side effects, which may not be cost‑effective for some patients due to the wide variability in relapse risk.10-12
Looking ahead, research focuses on new complement inhibitors, such as ravulizumab, crovalimab, and factor B inhibitors, with longer dosing intervals, costs, and improved safety. Advances in biomarkers and genetic profiling may also enable faster, personalized treatments, preventing unnecessary delays and complications.19

