











 Portugués (pdf)
Portugués (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUÇÃO
Phytophthora cinnamomi é um organismo da classe Oomiceta que está associado a grandes perdas económicas devido ao seu carácter fitopatogénico. É um dos parasitas de plantas mais devastadores conhecidos. Possui uma ampla distribuição a nível global e uma grande variedade de hospedeiros (Burgess et al., 2017). Além de causar perdas económicas substanciais na agricultura, silvicultura e horticultura, P. cinnamomi causa um enorme impacto nos ecossistemas naturais e na biodiversidade. Muitos estudos têm sido feitos ao nível do impacto que este organismo invasor tem na agricultura e nos ecossistemas através do reconhecimento de elementos génicos associados á infeção causada por P. cinnamomi (Zentmyer, 1980; Hardham, 2005). Neste trabalho o objetivo principal é a análise e caracterização de sequências depositadas em bancos de dados que codifiquem proteínas do metabolismo fundamental de P. cinnamomi. Os objetivos específicos desta pesquisa focam-se na utilização de ferramentas bioinformáticas a fim de obter informações relevantes sobre a localização subcelular, características físico-químicas e da estrutura 3D de proteínas expressas por genes de P. cinnamomi. É de fundamental importância a interpretação de informação biológica para a classificação e associação de características distintivas em todos os domínios da vida. Entende-se como metabolismo fundamental um variadíssimo conjunto de reações que de alguma forma são comuns a todas as formas de vida. Estas reações responsáveis pelos processos de síntese e degradação dos nutrientes na célula, permitindo o crescimento e reprodução das células, permite-nos compreender quais são respostas deste organismo às mudanças de ambiente para que possam ser desenvolvidas abordagens mais precisas no controle epidemia tão devastadora provocada pelo Oomiceta P. cinnamomi (Reeves, 1974; Ebenhöh, 2001; de Andrade Lourenço et al., 2020). A maioria das estruturas que compõem os seres vivos é fabricada a partir de três classes básicas de moléculas: aminoácidos, glícidos e lípidos. O metabolismo concentra-se na síntese destas moléculas ou na sua degradação para uso como fonte de energia e formação das estruturas celulares. Muitos compostos bioquímicos podem ser ligados entre si formando polímeros, como o DNA e proteínas (Devine et al., 1948). Estas macromoléculas são parte essencial de todos os organismos vivos. Para a correta caracterização da função de um determinado gene são usadas ferramentas bioinformáticas de análise de sequências genómicas de modo a inferir a sua função relacionando o grau de homologia entre sequências, os seus domínios e famílias com o produto proteico originado. Embora se conheça muito sobre o processo de infeção e o desenvolvimento da doença da tinta provocada por P. cinnamomi o conhecimento sobre os fatores (genes e proteínas) do metabolismo fundamental deste Oomiceta são pouco conhecidos, apesar de fundamentais para compreender o seu desenvolvimento e adaptação às alterações climáticas a fim de melhor estabelecer estratégias de controlo e combate.
MATERIAL E MÉTODOS
Bases de dados
A pesquisa de genes foi realizada através da base de dados NCBI (National Center for Biotechnology Information: https://www.ncbi.nlm.nih.gov/) onde foram usadas as sequências genómicas depositadas no primeiro contig LGSK01000001.1 pertencente a estirpe MP94-48 de P. cinnamomi.
Pesquisa de ORF’s
A ferramenta ORFfinder (https://www.ncbi.nlm.nih.gov/orffinder/), permitiu encontrar ORFs (as fases de leitura aberta ou parte codificante dos genes) no genoma de P. cinnamomi, utilizando como parâmetros de busca: sequencias até 300 nucleóticos, código genético standard e ORF usando codão de iniciação “ATG”. Sequências consideradas homologas são aquelas que apresentam uma percentagem de homologia superior a 80% de modo a ser possível afirmar que estão relacionadas ancestralmente. A ferramenta SmartBlast (https://blast.ncbi.nlm.nih.gov/smartblast/) utiliza um algoritmo heurístico de modo a produzir esses resultados usando uma combinação de uma pesquisa BLASTP otimizada, uma nova implementação do BLAST destinada a encontrar correspondências estreitamente relacionadas e um alinhamento múltiplo.
Localização subcelular das proteínas caracterizadas
A busca por um peptídeo sinal foi realizada com a ferramenta SignalP 5.0, que está disponível em http://www.cbs.dtu.dk/services/SignalP/index.php. Sinal peptídico (PS) são pequenas sequências de aminoácidos, que são clivadas durante a translocação da proteína através da membrana, permitindo prever a sua localização na célula (Emanuelsson et al., 2007). A possível localização subcelular foi confirmada pelas ferramentas Cello v.2.5, LOCTree3 e EukmPLoc, disponíveis em http://cello.life.nctu.edu.tw/, https://rostlab.org/services/loctree3/, http://www.csbio.sjtu.edu.cn/bioinf/euk-multi-2/ respetivamente (Yu et al., 2006; Chou e Shen, 2010; Goldberg et al., 2012).
Caracterização físico-química das proteínas
A caracterização físico-química foi obtida a partir da plataforma Expasy - ProtParam Server disponível em https://web.expasy.org/protparam/, e através desta análise foi possível obter informações importantes sobre a estrutura e propriedades das proteínas.
Os parâmetros calculados pelo ProtParam incluem peso molecular, ponto isoelétrico, composição de aminoácidos, índice de instabilidade, índice alifático e média de hidropaticidade (GRAVY). O índice de instabilidade fornece uma estimativa da estabilidade da proteína em um tubo de ensaio. É possível calcular um índice de instabilidade (II) que é definido como:
i = L-1
II = (10 / L) * ∑ DIWV (x [i] x [i + 1])
i = 1
onde: L é o comprimento da sequência, DIWV (x [i] x [i + 1]) é o valor do peso da instabilidade para o dipéptido que começa na posição i. Uma proteína cujo índice de instabilidade é menor que 40 é predita como estável, um valor acima de 40 prediz que a proteína pode ser instável. O índice alifático de uma proteína é definido como o volume relativo ocupado pelas cadeias laterais alifáticas (alanina, valina, isoleucina e leucina). Pode ser considerado como um fator positivo para o aumento da termo estabilidade de proteínas globulares. O índice alifático de uma proteína é calculado de acordo com a seguinte fórmula:
Índice alifático = X (Ala) + a * X (Val) + b * (X (Ile) + X (Leu))
Onde: X (Ala), X (Val), X (Ile) e X (Leu) é a percentagem molar (100 x mol) de alanina, valina, isoleucina e leucina. Os coeficientes a e b são o volume relativo da cadeia lateral da valina (a = 2,9) e das cadeias laterais Leu / Ile (b = 3,9) para a cadeia lateral da alanina (Ikai, 1980). O valor de GRAVY para um peptídeo ou proteína é calculado como a soma dos valores de hidropatia de todos os aminoácidos, divididos pelo número de resíduos na sequência e indica a solubilidade, com valores positivos (negativos) para proteínas hidrofóbicas (hidrofílicas) (Kyte e Doolittle, 1982).
A identificação de domínios foi realizada com a ferramenta Prosite disponível em https://prosite.expasy.org/, e a ferramenta Pfam - EMBL disponível em http://pfam.xfam.org/.
Estrutura 3D das proteínas
A estrutura 3D das proteínas foi obtida através do servidor Phyre2 disponível em http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index, e para a visualização das estruturas foi usado o programa PyMOL em http://www.pymol.org.
RESULTADOS E DISCUSSÃO
A análise da sequência genómica de P. cinnamomi depositada no banco de dados NCBI foi em primeira instância realizada com apoio da ferramenta bioinformática ORFfinder disponivel em https://www.ncbi.nlm.nih.gov/orffinder/, de forma a encontrar fases de leitura abertas que codifiquem proteínas relacionadas a fatores do metabolismo fundamental de P. cinnamomi. O termo ORF (open Reading frame) refere-se a uma parte de uma sequência nucleotídica que tem a potencialidade de codificar uma proteína, isto é, que contém um codão ou triplete de iniciação e um codão de terminação (Salinas et al., 2016). Após analisar as ORFs com o SmartBLAST (programa para comparação de consultas de proteínas ou DNA com bancos de dados de proteínas ou DNA em qualquer combinação) foram selecionadas ORFs que codificam proteínas que possivelmente estarão associadas a reações do metabolismo de P. cinnamomi. Os dados apresentados na tabela 1 são relativos aos genes e seus produtos, escolhidos após a identificação das ORFs e posterior comparação com SmartBLAST. A escolha foi baseada na relação entre o produto proteico, a percentagem de homologia entre sequências e a literatura científica disponível. A maioria das ORF’s codificam proteínas homólogas a proteínas do metabolismo de diferentes espécies do género Phytophthora especialmente P. infestans, P. megakarya, P. nicotianea e P. parasítica. As proteínas mais frequentes estão relacionadas com o metabolismo fundamental e proteínas que estão relacionadas com a patogenicidade destes organismos, embora exista um grande número de ORF’s que codifiquem para proteínas hipotéticas ou com função desconhecida. Os dados relativos a essa análise são apresentados no Quadro 1.
Quadro 1 Genes associados ao metabolismo fundamental de Phytophthora cinammomi. Indicam-se as referências do gene e da proteína deduzida nas bases de dados bem como a função da proteína e a percentagem de homologia no organismo respetivo
| GENE | NCBI | PROTEÍNA | FUNÇÃO | UNIPROT | ORGANISMO | COBERTURA/HOMOLOGIA |
| GQ600_10541 | KAF1794404 | Dioxigenase dependente de alfa-cetoglutarato- AlkB | Atividade da ubiquitina | A0A6A5JT99 | Phytophthora cactorum | 99% / 92.61% |
| PHMEG_000759 | OWZ18325 | Helicase dependente de ATP | Atividade de helicase | A0A225X269 | Phytophthora megakarya | 100% / 98.92% |
| PHMEG_00024779 | OWZ03481 | Glicerol quinase | Atividade da quinase | A0A225VD90 | Phytophthora megakarya | 98% / 78.38% |
| PHPALM_12227 | POM71226 | Poly(A) polimerase | Atividade de polinucleotídeo adenililtransferase | A0A2P4Y097 | Phytophthora palmivora var. palmivora | 100% / 87.15% |
| PITG_09292 | XP_002903577.1 | Dicer 1 (DCL1) | Atividade da ribonuclease III | D0NBC4 | Phytophthora infestans T30-4 | 100% / 80.75% |
| AM587_10017495 | KUF80413.1 | Pumilio 1 | Ligação ao RNA | A0A0W8C8Q2 | Phytophthora nicotianae | 99% / 86.85% |
| PPTG_04922 | XP_008895514.1 | Ribocinase | Atividade da ribocinase | W2R2M1 | Phytophthora parasitica (cepa INRA-310) | 90% / 87.93% |
| PITG_01564 | XP_002908209.1 | Aminoácido acetiltransferase | Atividade da N-acetiltransferase de L-glutamato | D0MTJ5 | Phytophthora infestans (strain T30-4) | 100% / 88.27% |
| AM587_10015544 | KUG00934.1 | Complexo de remodelação de cromatina da cadeia de ATPase | Ligação de DNA, ligação de ATP | A0A0W8DXS5 | Phytophthora nicotianae | 100% / 91.92% |
| PITG_10457 | XP_002902245.1 | RNA polimerase1 dependente de RNA (RDR1) | Ligação ATP, Atividade de polimerase de RNA 5'-3 | D0NFC8 | Phytophthora infestans (cepa T30-4) | 99% / 79.52% |
| F443_01087 | ETI56339.1 | Glicina-tRNA ligase | Ligação de ATP, atividade da glicina-tRNA ligase | V9FXY7 | Phytophthora parasitica P1569 | 99% / 96.47% |
A previsão da localização subcelular de proteínas fornece informações muito úteis acerca da sua possível função, pois a função de uma proteína está intimamente relacionada com sua localização (Chou et al., 2003). Os resultados do Quadro 2 foram obtidos através das ferramentas de localização celular e não se apresentaram de forma conclusiva sobre a real localização, pois foram bastante diferentes entre as três ferramentas. Cada uma destas ferramentas utiliza algoritmos diferentes para a apresentação de resultados, o que sugere a falta de experiências in vitro que determinem a sua real localização.
Quadro 2 Localização subcelular das proteínas deduzidas, utilizando as ferramentas SignalP, CELLO v.2.5, LOCTree3 e Euk-mPloc
| PROTEINA | SIGNALP | CELLO V.2.5/scoore | (LOCTREE3) | (EUK-MPLOC) |
| Dioxigenase dependente de alfa-cetoglutarato- AlkB | no | Citoplasma 0.468 | 84% Núcleo | Citoplasma. |
| Helicase dependente de ATP | no | Núcleo 0.620 | 90% Núcleo | Núcleo |
| Glicerol quinase | sim | Membrana plasmática 0.489 | 80% Peroxissoma | Citoplasma |
| Poly(A) polimerase | no | Cloroplasto 0.710 | 84% Núcleo | Núcleo |
| Dicer 1 (DCL1) | no | Citoplasma 0.917 | 82% Núcleo | Mitocondria, núcleo |
| Pumilio 1 | no | Núcleo 0.914 | 87% Citoplasma | Citoplasma |
| Ribocinase | no | Cloroplasto 0.777 | 84% Mitocondria | Citoplasma |
| Aminoácido acetiltransferase | no | Membrana plasmática 0.356 | 80% Cloroplasto | Cloroplasto |
| Complexo de remodelação de cromatina da cadeia de ATPase | no | Núcleo 0.530 | 87% Núcleo | Núcleo |
| RNA polimerase1 dependente de RNA (RDR1) | no | Membrana plasmática 0.544 | 82% Núcleo | Núcleo |
| Glicina-tRNA ligase | no | Citoplasma 0.797 | 86% Citoplasma | Citoplasma |
Os dados do Quadro 3 referem-se às características físico-químicas das proteínas selecionadas. Estes dados são importantes para um melhor entendimento das funções bioquímicas das proteínas e foram obtidos usando a ferramenta ProtParam que calcula várias propriedades físico-químicas que podem ser deduzidas a partir de uma sequência de proteínas. Nenhuma informação adicional é necessária sobre a proteína em consideração, a proteína pode ser especificada como um número de acesso ou ID SwissProt/TrEMBL, ou na forma de uma sequência bruta em formato FASTA.
Quadro 3 Caracterização físico-química (numero de aminoácidos, peso molecular, índices de estabilidade e alifático e hidropaticidade) das proteínas obtida através da ferramenta Expasy - ProtParam Server
| PROTEINA | Nº AA | PESO MOLECULAR | PI | INDICE DE INSTABILIDADE | ÍNDICE ALIFÁTICO | MÉDIA GERAL DE HIDROPATICIDADE ( GRAVY ) |
| Dioxigenase dependente de alfa-cetoglutarato- AlkB | 918 | 104925.48 | 6.19 | 45.88 (instável) | 81.35 | -0.354 |
| Helicase dependente de ATP | 1168 | 132488.90 | 8,65 | 50,49 (instável) | 77,65 | -0,592 |
| Glicerol quinase | 550 | 60170.84 | 7.70 | 39.51 (estável) | 94.18 | 0.013 |
| Poly(A) polimerase | 692 | 76653.46 | 7.90 | 55.96 (instável) | 78.64 | -0.379 |
| Dicer 1 (DCL1) | 1664 | 186186.12 | 5.44 | 42.86 (instável) | 86.17 | -0.334 |
| Pumilio 1 | 875 | 97313.93 | 6.23 | 56.22 (instável) | 65.11 | -0.605 |
| Ribocinase | 332 | 34602.58 | 4.84 | 28.16 (estável) | 97.83 | 0.214 |
| Aminoácido acetiltransferase | 567 | 61997.17 | 7.62 | 39.93 (estável) | 93.17 | -0.053 |
| Complexo de remodelação de cromatina da cadeia de ATPase | 1362 | 158231.05 | 8.68 | 45.04 (instável) | 75.93 | -0.819 |
| RNA polimerase1 dependente de RNA (RDR1) | 2680 | 302536.42 | 5.83 | 45.29 (instável) | 86.28 | -0.261 |
| Glicina-tRNA ligase | 681 | 76001.24 | 5.88 | 38.73 (estável) | 84.36 | -0.270 |
A identificação dos domínios e sítios ativos foi feita através da ferramenta PROSITE e os resultados são apresentados no Quadro 4. Através da análise dos resultados desta tabela foi possível deduzir a possível função das proteínas selecionadas com recurso á literatura científica disponibilizada nas bases de dados de proteínas Uniprot e Pfam-EMBL. A modelagem 3D das proteínas foi realizada através servidor Phyre2 e os resultados estão apresentados nas Figuras 1 e 2.
Quadro 4 Predição estrutural de proteínas usando Phyre2 e caracterização dos seus domínios e sítios activos usando o programa PROSITE
| PROTEINA | PHYRE2 | PROSITE | EMBL | |||
| Confiança | /Cobertura | Domínio | Sítios activos | Domínio | ||
| Dioxigenase dependente de alfa-cetoglutarato- AlkB | 100% | 50% | HECT, FE2OG_OXY | Glicil tioester intermediário | HECT, FE2OG_OXY | |
| Helicase dependente de ATP | 100% | 56% | S1, HELICASE_ATP_BIND_1, HELICASE_CTER | __ | S1, HELICASE_ATP_BIND_1, HELICASE_CTER | |
| Glicerol quinase | 100% | 88% | __ | __ | Família FGGY de carboidratos quinases, domínio N-terminal, Família FGGY de carboidratos quinases, domínio C-terminal | |
| Poly(A) polimerase | 100% | 72% | __ | __ | Domínio central da polimerase poli (A) Domínio de ligação ao RNA previsto em poli (A) polimerase | |
| Dicer 1 (DCL1) | 100% | 73% | DICER_DSRBF, RNASE_3_2, | __ | ||
| Pumilio 1 | 100% | 37% | PUM_HD | __ | __ | |
| Ribocinase | 100% | 92% | __ | __ | __ | |
| Aminoácido acetiltransferase | 100% | 74% | GNAT | __ | ||
| Complexo de remodelação de cromatina da cadeia de ATPase | 100% | 45% | HMG_BOX_2, HELICASE_ATP_BIND_1, HELICASE_CTER, SANT | N-terminal da família SNF2 Caixa de HMG; Caixa HMG (grupo de alta mobilidade) | ||
| RNA polimerase1 dependente de RNA (RDR1) | 100% | 28% | HELICASE_CTER, | __ | ||
| Glicina-tRNA ligase | 100% | 87% | WHEP-TRS, AA_TRNA_LIGASE_II | __ | ||
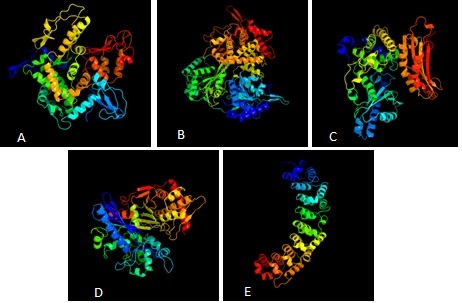
Figura 1 Representação da estrutura 3D das proteínas: A - Dioxigenase dependente de alfa-cetoglutarato- AlkB, B - Helicase dependente de ATP, C - Glicerol quinase, D - Poly(A) polimerase, E - Pumilio 1.
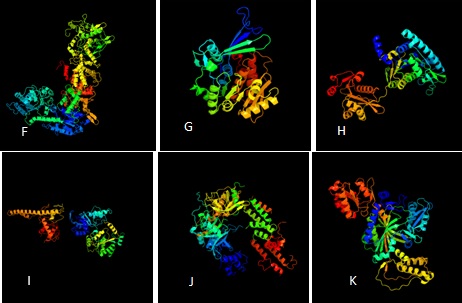
Figura 2 Representação da estrutura 3D das proteínas: F - Dicer 1 (DCL1), G - Ribocinase, H - Aminoácido acetiltransferase, I - Complexo de remodelação de cromatina da cadeia de ATPase, J - RNA polimerase1 dependente de RNA (RDR1), K - Glicina-tRNA ligase.
Dedução da função proteica através da interação dos domínios
A proteína dioxigenase dependente de alfa-cetoglutarato Tipo AlkB pertence à família das dioxigenases dependentes de Fe (II) / αKG com domínio HECT. Em particular, todas as dioxigenases da família AlkB usam um mecanismo de inversão de base para inverter a sua base alvo da hélice de DNA de fita dupla no seu sítio ativo e o núcleo do seu domínio catalítico contém uma dobra de β-hélice de fita dupla que é conservada entre dioxigenases dependentes de Fe (II) / αKG (Shen et al., 2014). O domínio HECT (homology para E 6-AP carboxyl terminus) é responsável pelo processo de ubiquitinação ao aceitar ubiquitina de uma enzima conjugadora de ubiquitina E2 na forma de um tioéster que é depois transferida para as proteínas alvo que são então degradadas no proteossoma 26S (Hatakeyama et al., 1997). Essas enzimas usam Fe (II) e 2-oxoglutarato como cofator para oxidar substratos orgânicos (Yu et al., 2006). Na reação AlkB, que requer ferro ferroso como cofator, o grupo metil é primeiro hidroxilado e depois espontaneamente libertado como formaldeído, regenerando a base normal. O oxigénio molecular é usado como o agente de oxidação, enquanto o co-substrato requerido 2-oxoglutarato (α-cetoglutarato) é convertido em succinato e 𝐶𝑂2 (Ougland et al., 2004). Esta proteína está envolvida na reparação do DNA e RNA através de mecanismos bioquímicos e moleculares e desta forma evita mutações ou modificações pós-transcripcionais e pós-traducionais atuando como um regulador proteico através da sinalização de proteínas indesejadas.
As helicases dependentes de ATP são enzimas que estão envolvidas em todos os processos do metabolismo do DNA e RNA. Estas proteínas estão divididas em 5 superfamílias (SF1-SF5) e todas elas se ligam ao ATP de forma a catalisar a remodelação de ácidos nucleicos. A helicase dependente de ATP descrita neste trabalho faz parte da família DEAD-box (superfamília SF2) com domínio S1 e C-terminal (Fairman-Williams et al., 2010). O domínio S1 caracteriza-se por se ligar ao RNA de forma a dar início à tradução e é encontrado numa grande diversidade de organismos (Bycroft et al., 1997), é essencial na tradução de proteínas, pois interage com o ribossoma e o mRNA e liga-se ao RNA de uma forma especifica na sequência. Tendo em consideração a mecânica da atividade da helicase da família DEAD-BOX, bem como da atividade bioquímica do domínio S1 e do domínio C-terminal (com função de determinar a especificidade á cadeia de RNA) poderá indica que esta proteína participa no processo de desnaturação da cadeia de ácidos ribonucleicos de uma forma dependente de energia tendo em consideração que a família DEAD-box apenas abrange helicases de RNA. A atividade bioquímica de definição central das proteínas da caixa DEAD traduz-se na capacidade de se ligar e hidrolisar o ATP em um ciclo estimulado pela ligação ao RNA de fita simples ou dupla (Jarmoskaite e Russell, 2012).
Glicerol quinase pertence à família FGGY com domínios N e C-terminais. Proteínas desta família são denominadas enzimas que catalisam a fosforilação de glicerol através da ligação do substrato de açúcar ao centro catalítico formando interações com o domínio N-terminal, o ATP liga-se junto à abertura entrando em contacto com os dois domínios alterando a conformação da estrutura proteica impedindo que o solvente entre no centro catalítico (Zhang et al., 2011). Poly(A) polimerase é a enzima responsável por adicionar uma cauda de poliadenina na extremidade 3’ de um transcrito de pré-mRNA. A sua adição é importante para a exportação nuclear, tradução e estabilidade do mRNA, evitando a sua degradação por ribonucleases. Esta proteína é composta por um domínio catalítico próximo ao terminal N e um domínio e uma região de ligação a RNA que se sobrepõe a um sinal de localização nuclear próximo ao terminal C (Martin et al., 2000). A proteína Dicer-1 faz parte da família das RNAases, contém um domínio de ligação ao RNA e caracterizam-se por atuar no processo de silenciamento de genes. A endoribonuclease Dicer-1 divide o RNA de fita dupla e o pré-microRNA em fragmentos curtos de RNA de fita dupla chamados RNA interferente curto e microRNA, respetivamente (Jaskiewicz e Filipowicz, 2008). É possível afirmar que esta proteína é muito importante no início do processo de silenciamento de genes por interferência de RNA.
A proteína de ligação ao RNA (Pumilio 1) descrita neste trabalho é membro da família PUF. As proteínas desta família são reguladores do desenvolvimento que controlam a estabilidade e a tradução do mRNA por sequências de ligação nas regiões 3 'não traduzidas de seus mRNAs alvo (Wang et al., 2002). São caracterizados pela presença de oito cópias em tandem de um motivo de sequência de 36 aminoácidos imperfeitamente repetido, a repetição Pumilio, cercado por uma região conservada N e C-terminal curta. As oito repetições e as regiões N e C-terminais formam o domínio de homologia de Pumilio (PUM-HD). O domínio PUM-HD é um domínio de ligação a RNA específico da sequência (Wang et al., 2001). O RNA liga-se à superfície côncava da molécula, onde cada uma das oito repetições da proteína faz contacto com uma base de RNA diferente através de três cadeias laterais de aminoácidos em posições conservadas (Wang et al., 2002).
A proteína Ribocinase descrita pertence à família pfkB e caracteriza-se por catalisar a fosforilação da D-ribose no O-5 formando D-ribose-5-fosfato numa reação que requer ATP e magnésio. O D-ribose-5- fosfato resultante pode então ser usado para a síntese de histidina e triptofano, ou como um componente da via da pentose fosfato. É o percursor essencial para a formação de nucleóticos (Sigrell et al., 1997). As N-acetiltransferases são enzimas que usam acetil coenzima A (CoA) para transferir um grupo acetil para um substrato, uma reação implicada em várias funções da resistência a antibióticos bacterianos e remodelação de cromatina. As N-acetiltransferases relacionadas a Gcn5 (GNAT) catalisam a transferência do acetil do doador CoA para uma amina primária do aceitador. As proteínas GNAT compartilham um domínio composto por quatro motivos de sequência conservados (Abboud et al., 2020). Apesar da diversidade de especificidades do substrato, os membros da superfamília GNAT demonstram notável similaridade na topologia de proteínas e no modo de ligação da acetil coenzima A, provavelmente refletindo um mecanismo catalítico conservado (Dyda et al., 2000). A proteína Aminoácido acetiltransferase é da família Gcn5 com domínio GNAT e e participa de processos do metabolismo de aminoácidos em P. cinnamomi.
A proteína ATPase do complexo remodelador de cromatina tem domínio Nterminal da família SNF2. O SNF2 funciona como o componente ATPase do complexo SNF2 / SWI, que utiliza energia derivada da hidrólise de ATP para interromper as interações histona-DNA, resultando no aumento da acessibilidade do DNA a fatores de transcrição como derivados de GAL4 ou a proteína de ligação a TATA-box (TBP), ao DNA associado a histona, embora a natureza exata dessa alteração estrutural não seja conhecida (Pazin e Kadonaga, 1997; Grüne et al., 2003). Devido à sua semelhança com o domínio de ligação ao c-Myb, foi sugerido que os domínios SANT são módulos de ligação ao DNA. O domínio SANT é um motivo de 50 aminoácidos presentes nas proteínas envolvidas na remodelação da cromatina e na regulação da transcrição. A maioria dos domínios SANT possui aminoácidos ácidos no início da hélice 2 e na hélice 3, enquanto os domínios de ligação ao DNA do tipo Myb têm resíduos carregados mais positivamente, em particular em sua terceira hélice de reconhecimento (Grüne et al., 2003). As proteínas com domínio HMG-box_2 parecem atuar principalmente como facilitadores arquitetónicos na montagem de complexos de núcleo-proteínas como por exemplo, na recombinação de ácidos nucleicos e no início da transcrição. As proteínas da HMG-box_2 podem ser direcionadas para locais específicos de DNA na cromatina por interações proteína-proteína ou pelo reconhecimento de estruturas específicas de DNA (Thomas e Travers, 2001). O domínio HELICASE_CTER está envolvido em processos de translocação de DNA. É necessário para acoplar a energia da hidrólise de ATP à atividade de remodelação do complexo SWI / SNF (Liu et al., 2010). As proteínas de remodelação da cromatina dependentes de ATP usam a energia liberada pela hidrólise de ATP para reposicionar nucleossomas em processos dependentes de DNA (Bansal et al., 2018). Esta ATPase, pode estar envolvida no complexo remodelador de cromatina sendo por isso um mecanismo associado ao metabolismo fundamental de P. cinnamomi.
O domínio catalítico central da enzima RDRP (RNA dependente de RNAPolimerase) é estruturalmente semelhante à subunidade beta das RNA polimerases dependentes de DNA (DDRP), no entanto os outros domínios do DDRP não mostram similaridade com os do RDRP. Por outro lado, esta enzima atua na amplificação de RNA de interferência em eucariontes, atuando de forma semelhante à RNA polimerase DNA dependente. Em adição pode-se afirmar que a RNA polimerase dependente de RNA, que atua na síntese de DNA a partir de um molde de RNA ao catalisarem a formação de ligações fosfodiéster entre ribonucleotídeos de uma maneira dependente do modelo de RNA (Ng et al., 2008). A proteína glicine-tRNA ligase pertence a família Aminoacil-tRNA sinteteases classe-II e é caracterizada por ser um grupo de enzimas que ativam aminoácidos e os transferem para moléculas específicas de tRNA como primeiro passo na biossíntese de proteínas. A presença de um domínio WHEP-TRS e um domínio de ligação ao anticodão exibem interações de alta afinidade com o tRNA aumentando assim a eficiência catalítica (Ray et al., 2011). A ligação de um aminoácido ao seu respetivo tRNA é levada a cabo por duas reações catalisadas por uma enzima apenas, uma aminoacil-tRNA-sintetase (também denominada de enzima de ativação), específica para cada aminoácido. A ativação do L-aminoácido pelo ATP com a formação de um aminoacil-AMP e formação de uma ligação éster do aminoácido com o tRNA, com formação do aminoacil-tRNA. Em reações catalisadas pelas aminoacil-tRNA sintetases classe II, o grupo aminoacil é acoplado ao 3'-hidroxil do tRNA (Wolf et al., 1999).
Todas estas enzimas que deduzimos a partir das sequencias genómicas depositadas nas bases de dados, helicases, poliApolimerases, aminoacil-tRNA sintetases participam nos mecanismos genéticos fundamentais desde a replicação, transcrição e tradução, mecanismos que garantem a transmissão da informação genética, a formação e estabilidade dos ácidos nucleicos e expressão da informação genética para a formação das proteínas que conferem as características que tornam este oomicete tão agressivo e letal para as plantas que parasita nomeadamente o Castanheiro.
CONCLUSÃO
As ferramentas bioinformáticas são muito importantes em pesquisas para a dedução de informação, a partir de sequências biológicas com o objetivo de caracterização de genes. Assim no presente trabalho foi possível prever através de homologia e similaridade de sequências entre proteínas a possível função de ORFs do genoma de P. cinnamomi. Mais estudos in sílico neste âmbito devem ser realizados para um melhor conhecimento acerca dos fatores genéticos que nos ajudam a compreender como este microrganismo vive, se reproduz e se adapta a mudanças no ambiente, pois apesar de nas bases de dados estar disponível todo o genoma sequenciado deste microrganismo, ainda é bastante reduzida a informação deduzida sobre o metabolismo deste fitopatogénio.

