










Services on Demand
Journal
Article
 Portuguese (pdf)
Portuguese (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkArquivos de Medicina
On-line version ISSN 2183-2447
Arq Med vol.25 no.1 Porto Feb. 2011
CASOS CLÍNICOS/ SÉRIE DE CASOS
Dermatomiosite na Criança – Caso Clínico
Dermatomyositis in ChilDren – CliniCal Case
Carla Dias1, Elisabete Moreira2, Cármen Lisboa2, Amélia Bartolo3, Irene Carvalho1, Iva Brito4, 5
1Serviço de Pediatria, Unidade Autónoma de Gestão da Mulher e da Criança do Hospital São João, Porto
2Serviço de Dermatologia, Unidade Autónoma de Gestão de Medicina do Hospital São João, Porto
3Serviço de Pediatria do Hospital São Sebastião, Santa Maria da Feira
4Unidade de Reumatologia Pediátrica, Unidade Autónoma de Gestão da Mulher e da Criança do Hospital São João, Porto
5Faculdade de Medicina, Universidade do Porto
RESUMO
A Dermatomiosite Juvenil é uma doença muscular inflamatória rara na infância, de natureza auto-imune e de etiologia desconhecida. Os critérios diagnósticos incluem manifestações clínicas musculo-esqueléticas e cutâneas, aumento das enzimas musculares, alterações na electromiografia e miosite na biópsia muscular. A ressonância magnética nuclear é útil na idade pediátrica para orientação diagnóstica. O tratamento de base é a corticoterapia. Outros imunosupressores (Metrotexato, Ciclosporina) também podem ser usados. O prognóstico da doença é favorável na maioria dos casos. Os autores descrevem o caso de uma criança de quatro anos com internamentos recorrentes por atingimento do estado geral e queixas músculo-esqueléticas inespecíficas. O aparecimento de lesões cutâneas a nível das mãos e cotovelos indiciaram o diagnóstico de Dermatomiosite Juvenil.
Palavras-chave: dermatomiosite, criança
ABSTRACT
Juvenile Dermatomyositis is an idiopathic inflammatory myopathy. it is a rare, autoimmune disorder in childhood, with unknown etiology. The diagnostic criteria consist of musculoskeletal and cutaneous manifestations, raised muscle enzymes, electromyographic abnormalities and myositis revealed by muscle biopsy. Mri is also an important diagnostic tool in pediatric patients. The treatment basis is corticotherapy. imunosupressant drugs such as methotrexate and cyclosporine may also be used. Prognosis is good in most patients. The case describes a four year-old child, who was recurrently admitted in hospital with constitutional and musculoskeletal symptoms and signs. The appearance of a typical skin rash on both hands and elbows allowed the diagnostic of probable Juvenile Dermatomyositis.
Key-words: dermatomyositis, child
Introdução
As miopatias inflamatórias idiopáticas são distúrbios raros em idade pediátrica. A Dermatomiosite Juvenil (DMJ) é a mais comum nesta faixa etária, com uma incidência de 3:1000000 casos/ano e predomínio no sexo feminino1. A idade média de diagnóstico é de sete anos, verificando-se 25% dos casos antes dos quatro anos de idade2. Difere da forma adulta pela maior incidência de vasculopatia e associação rara com doença maligna1,3.
Dados sobre a patogénese da DMJ sugerem tratarse de uma vasculopatia envolvendo mecanismos de imunidade celular contra antigénios musculares, imunocomplexos circulantes e acção do complemento. A sua etiologia poderá estar relacionada com factores infecciosos víricos (enterovírus) ou bacterianos (estreptococo do grupo A) assim como com factores genéticos (associação com os alelos HLA-DRB1*0301 e HLA-DQA1*0501)1. Polimorfismos genéticos da interleucina 1 (IL-1) parecem também ser promotores da doença4.
Os critérios diagnósticos de Bohan e Peter5 são consensuais e integram dados clínicos (fraqueza muscular proximal simétrica), lesões cutâneas típicas (eritema heliotrópico, sinal de Gottron), laboratoriais (aumento da enzimas musculares), electromiográficos (miopatia) e histológicos (miosite). O diagnóstico é definitivo, quando se associam três ou quatro critérios à presença das alterações cutâneas típicas, e provável se o doente preencher dois critérios na presença das alterações cutâneas.
Caso clínico
Os autores descrevem o caso de uma criança de sexo masculino, com quatro anos de idade, sem antecedentes pessoais ou familiares de relevo, que inicia queixas músculo-esqueléticas inespecíficas em Fevereiro 2005, sendo orientado para a Consulta de Pediatria do hospital da área de residência. Em Junho 2005 é internado nessa instituição por lesões da mucosa oral sugestivas de estomatite aftosa. Por manter queixas álgicas dos membros inferiores (MI), é medicado comum anti-inflamatório não esteroide (ibuprofeno) com melhoria sintomática. É novamente internado após um mês, por febre, anorexia, fadiga e mialgiados músculos proximais dos MI. Apresentava sinais de artrite do joelho esquerdo e punho direito e nódulos subcutâneos violáceos nos MI e cotovelos. Analiticamente refere-se anemia (hemoglobina:10,1g/dl com volume globular médio: 80fl), elevação da velocidade de sedimentação (VS: 47mm/1ªh), da proteína C-reactiva (PCR: 11mg/l) assim como das enzimas TGO (58U/l) e desidrogenase láctica (DHL: 356U/l). O estudo imunológico e asserologias (Borrelia burgdorferi, Citomegalovirus, vírus da hepatite B e C, Epstein Barr, Parvovirus B19 e Coxsackie) foram negativos. A ecografia abdominal mostrou hepatomegalia homogénea (12,9cm), sem outras alterações. A ecografia de partes moles e o ecocardiograma foram normais. A capilaroscopia mostrouáreas com poucos capilares curtos e desorganizados, sugestivo de doença do tecido conjuntivo, pelo que iniciou corticoterapia (prednisolona-1mg/Kg/dia), verificando-se melhoria sintomática significativa.
Durante o decréscimo da corticoterapia, é novamente internado por febre e recusa da marcha por coxalgia esquerda. A ressonância Magnética nuclear (RMN) da região pélvica revela alterações inflamatórias inespecíficas dos feixes musculares da raiz da coxa, com conservação da sua morfologia. Nessa altura, verificam-se úlceras cutâneas no cotovelo (Figura 1) e pavilhões auriculares, sendo transferido para o serviço de Pediatria do hospital São João para apoio de reumatologia e Dermatologia Pediátricas.

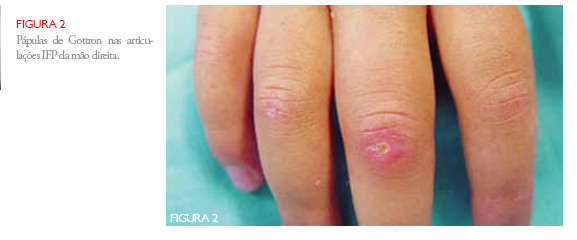
Na admissão apresentava razoável estado geral, úlceras cutâneas no cotovelo direito e pavilhões auriculares e pápulas eritematosas com descamação nas articulações interfalângicas (IF) proximais sugestivas de calcinose cutânea e pápulas de Gottron, respectivamente (Figura 2
). O exame músculo-esquelético mostrava diminuição da amplitude de movimentos dos cotovelos e ancas e fraqueza muscular proximal dos membros inferiores (grau II ) com sinal de Gower positivo. Analiticamente o hemograma era normal, sendo de destacar elevação daVS (85mm/1ªh), PCR (92mg/l) e das enzimas musculares séricas: TGO/ tGP (70/106U/l), DHL (320U/l), Creatinacinase (CK:51U/l) e Aldolase (16.5U/l). O estudo imunológico mostrou um aumento das imunoglobulinas igG e igM e o estudo de automunidade (anticorpos anti-nucleares, anti-ENA e Factor reumatoide) foi negativo. As radiografias das ancas, joelhos e punhos e a electromiografia ( do músculo tibial anterior não revelaram alterações. A biópsia cutânea mostrou dermatite com sinais de hemorragia antiga e estudo de imunofluorescência directa negativo.
Perante os achados clínicos e laboratoriais, presume-se o diagnóstico de DMJ e o doente reinicia corticoterapia (prednisolona 1mg/Kg/dia) com melhoria clínica e normalização dos parâmetros analíticos em quinze dias.
Seguido em Consulta de reumatologia pediátrica, manteve-se assintomático mas com uma dose mínima de corticoide.
Discussão
A sintomatologia na DMJ é predominantemente muscular e insidiosa, mas os sintomas constitucionais podem dominar o quadro clínico, o que geralmente atrasa o diagnóstico de seis a oito meses em média1. Neste caso, o quadro clínico inicial é consistente com a apresentação mais comum. Existe predomínio, ao longo de oito meses, das queixas músculo-esqueléticas tais como fraqueza, mialgia, artralgia e artrite (atinge até 30% dos doentes) em relação às queixas cutâneas. A presença no último internamento de lesões cutâneas de calcinose (presentes em 40 a 75% dos casos) indicia uma doença subjacente agressiva com evolução arrastada6. Não há, no entanto, atingimento de outros órgãos ou sistemas para além das clássicas alterações cutâneas e fraqueza muscular, apesar do atingimento multissistémico ser frequente na criança1,7,8.
Analiticamente, é observado o aumento característico das enzimas musculares séricas. O padrão é muito variável, sendo a CK a que menos se relaciona com a actividade da doença, podendo não haver elevação na fase aguda. As enzimas aldolase, DHL e TGO, apesar de menos específicas, reflectem melhor a actividade da doença, pelo que é importante a sua determinação e valorização precoces7. Anticorpos antinucleares podem estar presentes em mais de 80% dos casos; o factor reumatóide e restantes auto anticorpos estão raramente presentes1,7,8.
A capilaroscopia periungueal é um exame não invasivo que pode revelar alterações típicas como neste caso e auxiliar no diagnóstico e seguimento destes doentes9. Na admissão, o doente preenchia, para além da presença do rash típico (pápulas de Gottron), pelo menos dois critérios diagnósticos: o critério clínico (fraqueza muscular proximal dos MI) e o laboratorial (elevação das enzimas musculares). Não se objectivaram alterações na EMG, provavelmente pelo facto do doente ter efectuado corticoterapia em altas doses nos três meses anteriores. A RMN muscular, apesar de não integrar os critérios diagnósticos, é um instrumento válido e fiável na medição da inflamação muscular na DMJ10,11. Objectiva a presença de miosite nos casos em que não existe aumento das enzimas musculares1. É cada vez mais usada para orientação diagnóstica, já que a biópsia muscular, para além de ser um procedimento invasivo, pode ser negativa em até 20% dos casos no início da doença, dado que a inflamação muscular é habitualmente um processo focal7. Assim, numerosos casos de DMJ, tal como o que os autores apresentam, não preenchem o critério histológico de doença e são classificados como Dermatomiosite provável, em relação aos critérios de Bohan e Peter.
O tratamento consiste em terapêutica de suporte e supressão da actividade inflamatória com corticoterapia1. Os agentes modificadores de doença (Metotrexato, hidroxicloroquina, Ciclosporina) são usados na tentativa de reduzir o uso de corticoides ou quando estes não são suficientemente eficazes no controlo da doença, existindo já alguns autores que preconizam o uso de Metrotexato em primeira linha12. Os agentes biológicos tais como anticorpos monoclonais (rituximab) e anti-TNF ainda estão em estudo1,7,8. Com tratamento adequado, o prognóstico a longo prazo é bom, não se verificando sequelas funcionais, para a maioria dos doentes6,13. Lesões severas da pele (calcinose), sintomas neurológicos, gastrointestinais ou pulmonares estão associados a prognóstico mais reservado14. A precocidade de diagnóstico e o tratamento adequado são fundamentais, pois influenciam o prognóstico destes doentes2.
Os autores apresentam este caso pela raridade deste distúrbio na idade pediátrica. Realçam também a forma agressiva de manifestação cutânea da DMJ neste doente.
Referências
1. Cassidy J, Petty R. Textbook of Pediatric Rheumatology. 5th Ed 2005:407-41. [ Links ]
2. Pachman LM, Abbott K, Sinacore JM, Amoruso L, Dyer A, Lipton R, et al. Duration of illness is an important variable for untreated children with Juvenile Dermatomyositis. J Pediatrics 2006;148(2):247-53. [ Links ]
3. Compeyrot-Lacassagne S, Feldman BM. Inflamatory myopathy in children. Rheum Dis North Am 2007;33(3):525-53. [ Links ]
4. Mamyrova G, OHanlon TP, Sillers L, Malley K, James-Newton L, Parks CG, et al. Cytokine Gene Polymorphisms as Risk and Severity Factors for Juvenile Dermatomyositis. Arthritis Rheum 2008;58(12):3941-50. [ Links ]
5. Bohan A, Peter JB. Polimyositis and Dermatomyositis. N Eng J Med 1975;292:344-403. [ Links ]
6. Sallum AME, Kiss MHB, Sachetti S, Resende MBD, Moutinho KC, Carvalho MS et al. Juvenile dermatomyositis: clinical, laboratorial, histological, therapeutical and evolutive parameters of 35 patients. Arq Neuro-Psiquiatr 2002;60:889-99. [ Links ]
7. Wedderburn L, Li C. Paediatric idiopathic inflamatory muscle disease. Best Pract Res Clin Rheumatol 2004;18 (3):345-58. [ Links ]
8. Pilkington C, Wedderburn L. Paediatric idiopathic inflamatory muscle disease - recognition and management. Drugs 2005;65(10):1355-65. [ Links ]
9. Nascif AK, Terreri MT, Len CA, Andrade LE , Hilário MO. Inflammatory myopathies in childhood: correlation between nailfold capillaroscopy findings and clinical and laboratory data. J Pediatr (Rio J) 2006;82(1):40-5. [ Links ]
10. Hilário MO, Yamashita H, Lutti D, Len C, Terreri MT, Lederman H. Juvenile idiopathic inflammatory myopathies: the value of magnetic resonance imaging in the detection of muscle involvement. Sao Paulo Med J 2000;118(2):35-40. [ Links ]
11. Maillard SM, Jones R, Owens C, Pilkington C, Woo P, Wedderburn LR , et al. Quantitative assessment of MRI T2 relaxation time of thigh muscules in Juvenile Dermatomyositis. Rheumatol 2004;43:603-8. [ Links ]
12. Tournadre A, Dubost JJ, Soubrier M. Comment traiter une myopathie inflammatoire de ladulte? Rev Rhum 2010;77(4):333-337. [ Links ]
13. Huber AM, Lang B, LeBlanc CM, Birdi N, Bolaria RK, Malleson P, et al. Medium and long-term functional outcomes in a multicenter cohort of children with Juvenile Dermatomyositis. Arthritis Rheum 2000;43:541-9. [ Links ]
14. Bowyer SL , Blane CE, Sullivan DB, Cassidy JT. Childhood Dermatomyositis: factors predicting functional outcome and development of dystrophic calcification. J Pediatr 1983;103(6):882-6. [ Links ]
Carla Dias Serviço de Pediatria, Unidade Autónoma de Gestão da Mulher e da Criança, Hospital São João Al. Prof. Hernâni Monteiro 4200-319 Porto. Email: carlajdias@gmail.com

