










Serviços Personalizados
Journal

Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
 Citado por SciELO
Citado por SciELO Links relacionados
 Similares em
SciELO
Similares em
SciELO Compartilhar

 Permalink
PermalinkArquivos de Medicina
versão On-line ISSN 2183-2447
Arq Med vol.28 no.1 Porto fev. 2014
ARTIGO DE REVISÃO
A influência do sistema nervoso Autónomo na resposta inflamatória da sepsis
The influence of the Autonomic nervous system in the inflammatory response of sepsis
João Gonçalves1
1Faculdade de Medicina Universidade do Porto
RESUMO
A Sepsis, síndrome de resposta inflamatória sistémica associada a infeção, é uma entidade clínica com crescente impacto social e médico. A desregulação da resposta inflamatória parece estar na base da sua fisiopatologia. O Sistema Nervoso Autónomo (SNA) assume um papel regulador de grande parte das funções orgânicas. Nos últimos anos, têm-se acumulado evidência de que a resposta imune está também sob a influência do SNA através dos seus dois braços eferentes: o sistema nervoso simpático (SNAS) e parassimpático (SNAP). A influência do SNAS parece ser complexa, apresentando efeitos pró e anti-inflamatórias, enquanto o SNAPS induz efeitos predominantemente anti-inflamatórios. Curiosamente, parece que estes sistemas atuam de forma sinérgica na regulação de resposta imune a nível do baço. A capacidade de o SNA modular a resposta inflamatória poderá levar ao surgimento de novas abordagens terapêuticas da Sepsis, como o recurso a agonistas ß2 ou dos recetores nicotínicos do tipo a7. Estas abordagens deverão ter em conta a importância que a estimulação do SNAS tem na otimização da pressão de perfusão tecidular nos doentes sépticos. Assim, o aprofundamento do conhecimento do papel do SNA na fisiopatologia da Sepsis poderá abrir novas perspetivas de tratamento desta entidade.
Palavras-chave: Sepsis, inflamação, sistema nervoso autónomo
ABSTRACT
Sepsis, systemic inflammatory response syndrome associated with infection, is a clinical entity with increasing social and medical impact. The deregulation of the inflammatory response seems to be in the basis of its pathophysiology. Autonomic Nervous System (ANS) assumes a regulatory role in the majority of the bodys functions. In the last years increasing evidence has shown that immune response is under influence of ANS through its two efferent arms: sympathetic (SNS) and parasympathetic nervous systems (PSNS). The influence of SNS seems complex showing pro and anti-inflammatory effects while PSNS has mainly anti-inflammatory features. Interestingly it seems that these systems act in a synergic way in the regulation of immune response at the spleen. ANSs ability to modulate the inflammatory response may lead to the emergence of new approaches to the treatment of Sepsis, like ß2 adrenergic receptor and a7 nicotinic receptors agonists. These approaches ought to respect the importance of the SNS stimulation to the optimization of perfusion pressure in septic patients. in this way the deeper understanding of ANSs role in the pathophysiology of Sepsis will open new perspectives regarding the treatment of this entity.
Key-words: Sepsis, inflammation, autonomic nervous system
Introdução
A Sepsis é uma entidade clínica que se define como uma Síndrome de resposta inflamatória Sistémica (SIRS) associada a infeção confirmada ou presumida.1 Trata-se de uma síndrome com significativo impacto social, atingindo cerca de 650.000 pessoas e sendo responsável pela morte de cerca de 200.000 pessoas anualmente nos EUA.2 Na verdade, a patogénese da Sepsis é extremamente complexa. Partindo da sua definição torna-se percetível que se trata de uma entidade clinica em que a reação inflamatória assume um papel central. Atualmente, acredita-se que a resposta a um agente agressor leva à produção de citocinas inflamatórias, transformando um fenómeno inflamatório localizado num evento sistémico – o SIRS.3 Embora o SriS seja um componente determinante nas fases iniciais da Sepsis, cada vez mais se consegue que o doente sobreviva a esta fase inflamatória inicial da Sepsis através da instituição precoce de antibioterapia, otimização da oxigenação tecidular e controlo do foco infecioso.4 todavia, os doentes que não atingem a cura nesta primeira fase, podem evoluir para uma fase mais tardia caracterizada por uma acrescida suscetibilidade a infeções secundárias, que poderão ser a causa da morte do doente. Assim, surgiu o conceito de imunoparalisia, situação anormal resultante do exagero da resposta anti-inflamatória compensatória (CARS).5,6 O Sistema Nervoso Autónomo (SNA) é constituído por dois componentes clássicos: o Sistema Nervoso Simpático (SNAS) e Sistema Nervoso Parassimpático (SNAPS).7 O SNA assume uma função de manutenção da homeostasia, influenciando desde a função cardiovascular, respiratória e gastrointestinal até ao controlo dos níveis glicémicos e a temperatura corporal.8 Nas últimas décadas têm-se verificado que a influência do SNA também se estende ao sistema imunológico. Embora classicamente o Sistema imunológico seja considerado como um sistema autónomo, fora do alcance do sistema nervoso, tal conceito foi desafiado à medida que se verificou que o Sistema Nervoso Central (SNC) possuía capacidade de modular a resposta inflamatória/imune, primeiro através do eixo Hipotálamo-Hipófise-Supra renal (HHSR) e seguidamente através do SNA.9 Tratando-se de uma entidade clinica que apresenta um sério desafio à manutenção da homeostasia corporal, a Sepsis induz profundas alterações no padrão de ativação do SNA.10 Ao impacto produzido pela patologia per se acrescentam-se as alterações induzidas pelas intervenções terapêuticas, nomeadamente o recurso a aminas para otimização das pressões de perfusão.8
Portanto, considerando que a Sepsis induz profundas alterações na padrão de ativação do SNA e tendo em conta o carácter inflamatório desta patologia, faz todo o sentido tentar compreender qual o papel do SNA na regulação da inflamação durante a Sepsis.
A ligação entre a inflamação e SNA
Sendo que o SNA é um sistema regulador de múltiplas funções orgânicas e uma vez que se tem descoberto interações entre o SNA e o sistema imune, é lógico equacionar a hipótese do SNA exercer funções reguladoras sobre a resposta inflamatória. Assim, para que o SNA assuma este papel, é necessário que exista: uma via aferente, que forneça informação sobre estado inflamatório; um centro regulador, que consoante as informações veiculadas gere uma resposta adequada às necessidades do organismo; uma via eferente que transmita essa resposta de modo a modular a resposta imune.11
Considerando que os centros reguladores das funções orgânicas do SA se encontram no SNC é lógico presumir que o mesmo se aplique para a função imune. Assim, existem dois modos de serem veiculadas informações ao SNC: a via humoral, por substâncias solúveis que ativem recetores a nível do SNC; a via hard-wired, em que as informações ao SNC são veiculadas por fibras nervosas.7,12
Para o papel de agente que atue pela via humoral surgem as citocinas, nomeadamente a IL-1 e o TNFα. Na verdade, como estas são geradas em situações de inflamação, servem, de uma forma simplista, como marcadores do estado inflamatório. Apesar da incapacidade da IL-1 ultrapassar a barreira hemato-encefálica (BHE), esta pode influenciar o SNC através de estruturas desprovidas de BHE, como os órgãos paraventriculares, ou através de recetores nas células endoteliais, que são responsáveis pela produção de mediadores secundários, como as prostaglandinas. O mecanismo da febre é um exemplo, já que a IL-1 induz alterações do termóstato hipotalâmico através da produção secundária de PGE2. 12,13
Por outro lado, sabe-se que a administração intra peritoneal de baixas doses de IL-1, insuficientes para elevar as concentrações plasmáticas de stacitocina, é capaz de induzir uma ativação do SNAS. Para além disto, num ensaio semelhante verificou-se que a administração intraperitoneal de IL-1 levava a um aumento da frequência de despolarizações do nervo vago.12 Assim, parece que informações relativas ao estado inflamatório sistémico podem serveiculadas ao SNC, via nervo vago, gerando uma resposta reflexa através do SNA – o reflexo inflamatório.9 As células glómicas presentes no corpo carotídeo parecem ser sensíveis ao lPS assim como à IL-1, Il-6 e TNFα.14 A estimulação destas células com LPS e TNFα é capaz de modular a sensibilidade do carpo carotídeo a estímulos como a hipoxia.15 Assim, a alteração da função desta estrutura pode servir como um meio alternativo de veicular informações sobre estado inflamatório ao SNC.
A via eferente do SNA por este reflexo inflamatório passará pelos seus dois componentes: o SNAS e o SNAPS. Os efeitos destes sistemas sobre a resposta inflamatória/imune serão abordados em maior pormenor nos dois capítulos seguintes.
O sistema simpático e a inflamação
O SNAS e o eixo HHSR são os dois sistemas, com origem no SNC, que classicamente são ativados em resposta a agentes de stress. Tem-se verificado que após eventos indutores de stress (quer físicos quer psicológicos) se gera um estado de imunossupressão. Na realidade, o cortisol, produzido pelas glândulas supra-renais após a ativação do eixo HHSR, parece ser o principal responsável por esta imunossupressão.5 Contudo, nas últimas décadas têm-se acumulado evidência que aponta para um papel importante do SNAS.
A Sepsis induz uma ativação precoce do SNAS, com uma produção e libertação intensificada dos seus principais mediadores: adrenalina e noradrenalina.8 Para o tratamento do choque séptico, é muito comum o uso de catecolaminas como objetivo de maximizar a pressão de perfusão dos órgãos.4 Assim, é muito importante perceber qual o impacto que este influxo de catecolaminas, endógenas e exógenas, tem na resposta imune/inflamatória no contexto da Sepsis.
Para que se possa compreender como é que o SNAS modula a resposta inflamatória é necessário primeiro reconhecer que o sistema imune é extremamente complexo, envolvendo uma cascata de células e moléculas mediadoras de grande diversidade. Deste modo, para simplificar a compreensão do papel do SNAS pode-se subdividir a resposta imune em várias fases. Contudo, deve-se ter sempre presente que estas fases não são independentes mas desenvolvem-se conjuntamente.
Imunidade inata
Reconhecimento da ameaça - TLR
Quando ocorre a invasão por um agente patogénico, como por exemplo uma bactéria, para que possa ser montada uma resposta há que, em primeiro lugar, identificar o agente agressor. tal acontece através de recetores que reconhecem padrões moleculares associados a agentes patogénicos (PAMPs). O lipopolissacarideo (LPS), existente nas bactérias Gram -é um exemplo de PAMP. Fazem parte desta classe de recetores várias famílias, como por exemplo os recetores Toll-like (TLR), que quando ativados iniciam a cascata inflamatória necessária à eliminação do agente agressor.16 Para a iniciação desta cascata é essencial o fator de transcrição NFKB. No estado basal, o NFKB encontra-se localizado no citosol, sendo que a estimulação dos tlr leva à sua ativação e translocação para o núcleo, onde promove a transcrição de genes envolvidos na resposta inflamatória, como as citocinas TNFα e IL-1.17
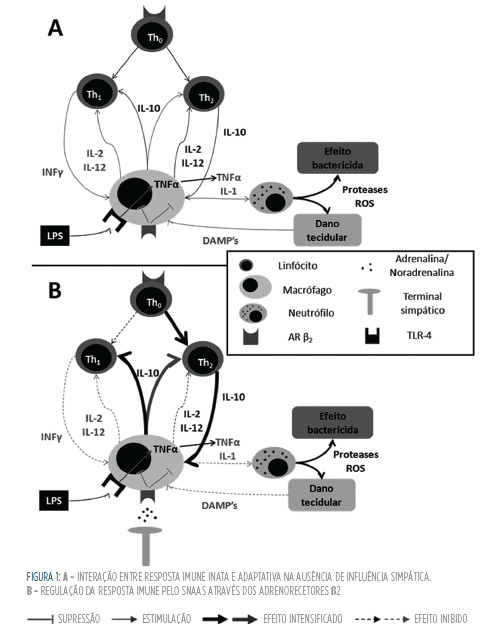
Acredita-se que a hiperestimulação dos TLR por antigénios patogénicos pode estar implicado na génese do estado hiperinflamatório associado à Sepsis.3 O SNAS é capaz de modular a ativação destes recetores. Demonstrou-se que adicionando noradrenalina a culturas de células mononucleares periféricas estimuladas com LPS, ocorre redução da produção de IL-1 e TNFα.18 Na verdade, este efeito é mediado via recetores adrenérgicos ß2, que medeiam grande parte dos efeitos do SNAS a nível da resposta imune.18,19 Partindo desta descoberta, vários estudos confirmaram que o recurso a agonistas β não-seletivos (como a isoprenalina)20 e β2-selectivos (como a terbutalina) induz uma resposta anti-inflamatória, com redução da produção de TNFα, IL-1e também da proteína high-mobility-group-box-1 (HmGB1), que funciona como uma citocina de fase tardia na Sepsis. Curiosamente, a estimulação ß1 parece aumentar a resposta pro-inflamatória e não suprimi-la.21 Por outro lado, polimorfismos do gene do recetor β2, caracterizados por menor capacidade de supressão da produção de IL-6, estão associados a pior prognóstico de doentes com choque séptico.22
Em contraponto com estes achados, verificou-se que a co-estimulação, de células de Kupffer e algumas linhas de macrófagos peritoneais, com lPS e noradrenalina leva não a uma redução da produção de citocinas, mas sim a um aumento sinérgico.18,19,23 Este efeito é mediado por recetores 24,25 adrenérgicos α2 (Arα2), especificamente α2A. 24,25 Uma importante fonte de noradrenalina é o trato gastrintestinal, sendo que durante a Sepsis a concentração de noradrenalina na circulação portal pode ser duas vezes superior à da circulação sistémica. A ativação de recetores α2 presentes nas células de Kupffer, através da ação da noradrenlina de origem entérica, leva a um aumento da produção de TNFα, IL-1 e IL-10 e parece estar associada ao surgimento de disfunção hepática em modelos animais da Sepsis.23,26,27
A evidência parece demonstrar que os monócitos circulantes apresentam um perfil de resposta β2, mas à medida que amadurecem e se deslocam para os tecidos perdem essa característica, passando a predominar os efeitos pró-inflamatórios α2A.18 Para além disto, é possível que a Sepsis induza uma alteração fenotípica das células de Kupffer caracterizada por upregulation dos recetores adrenérgicos α2A.25 Deste modo, as catecolaminas poderão suprimir a resposta inflamatória nas células circulantes ao mesmo tempo que estimulam a resposta inflamatória local. Este perfil parece indicar que o SNAS poderá ter um papel importante na teoria da Compartimentação da resposta inflamatória da Sepsis.28 todavia, estes dados complicam a interpretação do efeito global do SNAS, indicando que poderá ter um papel complexo e variável.
Quimiotaxia e resposta local
Após o reconhecimento da invasão, ocorre a ativação das células imunes locais, como os mastócitos e os macrófagos tecidulares, sendo que estas células produzem várias substâncias (citocinas, quimiocinas e mediadores lipídicos) vitais ao recrutamento de células imunes competentes capazes de gerar uma resposta inflamatória. Assim, a secreção de citocinas, como a IL-1 e o TNF-α, e quimiocinas, como a IL-8 e CCL-3, induz o recrutamento de neutrófilos, monócitos e linfócitos. Neste ponto, o SNAS produz efeitos mais complexos. Por um lado, poderá aumentar a produção de IL-829 mas, por outro lado, verificou-se que as catecolaminas suprimem a produção de CCl3.18,30 Para além disto, a supressão da produção de IL-1 e TNFα induziria uma redução da quimiotaxia. Sabe-se também que as catecolaminas possuem efeitos supressores da atividade dos neutrófilos, reduzindo o burst oxidativo, fagocitose e quimiotaxia, sendo estes mediados por recetores β.18 A estimulação a-adrenérgica tende a causar o efeito inverso.19 todavia a modulação da produção de quimiocinas e do recrutamento celular possui duas facetas opostas. Por um lado, a supressão da sua produção causará um efeito anti-inflamatório mas, por outro lado, poderá reduzir o clearance bacteriano, o que por si só poderá prolongar a resposta inflamatória.
Imunidade adquirida
A resposta adaptativa - Equilíbrio TH1/TH2:
Células apresentadoras de antigénios (APC)
Embora se dê particular atenção à desregulação do sistema imune inato, ocorrem também alterações na resposta imune adaptativa durante a Sepsis. Para a construção de uma resposta imune adquirida, assumem um papel central as APC, que internalizam, processam e apresentam os antigénios às células TH. São consideradas APC: as células dendríticas, os macrófagos e os linfócitos B. Da interação entre as APC e as células TH gera-se a ativação dos mecanismos de imunidade adquirida, quer sejam eles celulares (ativação macrofágica e de células t citotóxicas) quer humorais (por produção de anticorpos). A partir a desta interação gera-se uma alteração fenotípica das células Th seguindo um fenótipo Th1 ou Th2. O fenótipo Th1 é caracterizado pela produção de INFγ, TNFα e IL-13, estando associado à ativação da imunidade celular e a um status imunológico pró-inflamatório. O perfil Th2 está associado à produção de IL-10, IL-4 e IL-3, induzindo um estado anti-inflamatório, e ativando a imunidade humoral (principalmente a produção da fração IgE). Crê-se que a capacidade de resposta imunológica depende muito do equilíbrio existente 18,31 entre as vias Th1/Th2.
Considerando que a Sepsis resulta de uma ativação inflamatória sistémica seria de esperar que a razão Th1/Th2 estivesse aumentada. Apesar disso para uma parte importante dos doentes sépticos o componente CARS é predominante, surgindo um estado de imunossupressão associado a uma redução da razão Th1/Th2.6
O SNAS induz profundas alterações no equilíbrio Th1/Th2, quer a nível das APC quer a nível linfocitário. Nas células dendríticas, a produção de IL-12 e INFγ é suprimida pela ação da noradrenalina e adrenalina, via recetores β2, inclinando o equilíbrio para uma resposta Th2.18,32 Como já foi referido acima, os efeitos nos monócitos/ macrófagos são mais complexos. Assim, nos monócitos, e em algumas linhagens de macrófagos, as catecolaminas suprimem a produção de IL-1 e TNFα,20,33 citocinas que estimulam os macrófagos a produzir IL-12, uma citocina tipicamente Th1. Para além disto, as catecolaminas também induzem um aumento da produção de IL-10, uma citocina que suprime as respostas Th1.34 Assim, através das APC, o SNAS parece favorecer uma resposta Th2.
Linfócitos
Os linfócitos apresentam diferentes expressões de recetores β-adrenérgicos à sua superfície. As células Th1 e os linfócitos B exprimem estes recetores ao passo que as células Th2 não.18 No que respeita ao eixo Th1 as catecolaminas parecem induzir um efeito puramente supressor: inibem a diferenciação de células Thnaive em Th119; reduzem a capacidade de proliferação das células Th1 podendo até induzir apoptose, possivelmente através da redução de recetores de il-2; reduzem a produção de citocinas Th1, como o INFγ.18 Aparentemente, no que respeita aos linfócitos Th2, as catecolaminas não induzem alterações diretas na função. Resumindo as catecolaminas induzem um efeito supressor sobre os linfócitos Th1, um efeito supressor sobre as citocinas Th1 produzidas pelas APC (por exemplo IL-12) e um efeito nulo sobre as células Th2. Assim, no que respeita à imunidade adquirida, as catecolaminas inclinam a resposta imune para um perfil Th2, caracterizada pela produção de citocinas inibidoras da resposta inflamatória.18
SNAS e as bactérias
Como já foi referido, o estudo do impacto do SNAS na resposta inflamatória da Sepsis é uma tarefa complicada devido ao grande número de parâmetros a considerar. Na verdade, os pontos previamente mencionados relatam apenas alterações na função das células do hospedeiro. Contudo, a Sepsis resulta da interação de um agente patogénico e da resposta gerada pelo hospedeiro. Assim, para se compreender o impacto que o SNAS tem na resposta inflamatória é preciso compreender também o seu impacto nos agentes patogénicos.35 Na realidade, vários estudos apontam para o facto de as catecolaminas terem a capacidade de induzir aumento da proliferação bacteriana e possivelmente um aumento da sua virulência.36 Para além deste efeito, parece que a modulação que o SNAS exerce sobre as células do sistema imune pode afetar o clearance bacteriano de forma negativa para um tipo bacteriano mas de forma positiva para outros tipos. De facto, quando se avaliou o clearance bacteriano em ratinhos sujeitos a simpatectomia, foi verificado que ocorria uma redução da disseminação bacteriana perante a infeção por Pseudomonas aeruginosa e um aumento da disseminação aquando de uma infeção por Staphylococcus aureus.37 Curiosamente, este estudo parece apontar para que o efeito do SNAS na imunidade seja também dependente do tipo de agente patogénico, possivelmente favorecendo o combate a infeções por Gram + e dificultando a eliminação de agentes Gram -. Tal poderá dever-se ao facto de estes agentes levarem à ativação de respostas imunes com perfis diferentes.
Recetores β e dessensibilização
A expressão dos recetores adrenérgicos nas membranas celulares é um processo dinâmico. De facto, a exposição prolongada a catecolaminas induz fenómenos de downregulation dos recetores β, envolvendo a fosforilação do recetor e consequente sequestro intracelular envolvendo β-arrestinas. Na verdade, um estudo que avaliava o impacto da isoprenalina na produção de TNFα por macrófagos CD14+,mostrou que, nas células obtidas de voluntários saudáveis, a isoprenalina induzia uma redução da produção desta citocina, sendo que este efeito não se verificou em macrófagos obtidos de doentes sépticos.38 Contudo, a via de sinalização dos recetores β estava intacta, pois a estimulação da enzima adenilciclase simulava os efeitos da isoprenalina.
Compreende-se que a modulação que o SNAS induz no sistema imunológico é um fenómeno complexo. Por um lado, parece induzir fenómenos predominantemente anti-inflamatórios com depressão quer da imunidade inata quer adquirida, por outro, poderá prolongar a resposta inflamatória devido a uma redução da capacidade de clearance bacteriano e da estimulação de recetores α. Acrescenta-se ainda o facto de o nível de ativação do SNAS poder estar sujeito a variações temporais ao longo do processo evolutivo da Sepsis. A Fig 1 esquematiza o papel na regulação da resposta imune.
O sistema parassimpático e A inflamação
O estudo do impacto do SNAPS na inflamação é mais recente e menos aprofundado que o estudo do SNAS. Contudo o volume de evidência a suportar o potencial clínico da modulação do SNAPS tem vindo a crescer de forma continua.
O interesse do SNAPS como sistema modulador da resposta inflamatória adveio da descoberta que a estimulação de macrófagos com acetilcolina inibe a produção de várias citocinas pró-inflamatórias (TNFα, IL-1, IL-6 e IL-18) sem contudo alterar a produção de IL-10.9 Posteriormente, verificou-se que a estimulação do nervo vago era capaz de reduzir a produção de várias citocinas inflamatórias (TNFα, IL-1, HMGB-1) em vários modelos de Sepsis.
A acetilcolina exerce as suas ações através da estimulação dos recetores nicotínicos (nAcCor) e muscarínicos (mAcCor). Contudo, a subunidade responsável pelo efeito anti-inflamatório é a subunidade α7 do recetor nicotínico da acetilcolina (a7nAcCor). Apenas a estimulação deste subtipo gera uma resposta anti-inflamatória relevante, pois a estimulação de nAcCor com outro tipo de subunidades ou a administração de agonistas muscarínicos periféricos não reduzem a resposta inflamatória induzida pela administração de lPS in vitro ou in vivo.39 Para além disto, o bloqueio do a7nAcCor anula a capacidade anti-inflamatória do nervo vago.40
Como foi dito acima, as fibras ascendentes vagais fazem parte da via hard-wired, que informa o SNC do estado inflamatório dos tecidos.7 No entanto, estes estudos mostraram que as fibras descendentes vagais poderão ser um dos componentes do braço eferente do SNC responsável por modular a resposta inflamatória.41
Para além da estimulação do nervo vago, os agonistasdo α7nAcCoR,como por exemplo a nicotina, colina e o GtS-21, assim como os inibidores da aceticolinesterase (IACE), têm capacidade de reduzir a produção de citocinas pró-inflamatórias, assim como melhorar a sobrevida nos modelos animais de Sepsis.41
Verificou-se que embora os mAcCor periféricos não induzam alterações da produção de citocinas o mesmo não se aplica aos mAcCor centrais. De facto, a estimulação dosrecetoresm1centrais ou então estratégias que aumentem a concentração de acetilcolina na fenda sináptica a nível central (antagonistas m2 ou iACE de ação central), induzem uma redução das concentrações plasmáticas de TNFα.39,42 Na verdade, este efeito pode dever-se à estimulação vagalpoisousodeagonistasm1levaaumaumento da frequência de despolarizações deste nervo.39
O baço parece ser um órgão indispensável para o funcionamento deste sistema colinérgico antiinflamatório. De facto, a esplenectomia anula os efeitos da estimulação vagal e dos agonistas α7nAcCoR.43 Curiosamente, o efeito da esplenectomia pode ser reproduzido causando apenas a ablação do nervo esplénico.44 Estes resultados são intrigantes porque nunca foram localizadas fibras colinérgicas no baço e o nervo esplénico é constituído puramente por fibras simpáticas.41 Assim, a estimulação do nervo vago nunca poderia modular diretamente a atividade das células imunes do baço. A presença de recetores α7nAcCoR nos neurónios pós-ganglionares simpáticos do gânglio celíaco pode explicar este ponto: a estimulação vagal liberta acetilcolina que, via α7nAcCoR, ativa neurónios simpáticos pós-ganglionares no gânglio celíaco.41,45 Seguidamente as fibras destes neurónios seguem no nervo esplénico e podem modular a função das células imunes do baço via recetores adrenérgicos.41 Como foi dito acima, os recetores β2 são responsáveis por muitos dos efeitos anti-inflamatórios das catecolaminas.19 Assim, acredita-se que a estimulação vagal produz um efeito anti-inflamatório por aumento da libertação de catecolaminas no baço e consequente estimulação de recetores β2.41 De facto, o antagonismo de recetores β246 ou a depleção de catecolaminérgica44 bloqueia o efeito anti-inflamatório da estimulação vagal. Este mecanismo de cooperação entre o SNAS e o SNAPS é curioso visto que estes dois sistemas funcionam de forma oposta na regulação da maior parte das funções orgânicas.7
Contudo, o funcionamento desta via anti-inflamatória colinérgica é ainda mais complexo. Como foi dito previamente, o baço não é enervado por fibras colinérgicas, contudo a estimulação do nervo esplénico induz um aumento das concentrações de acetilcolina no baço. Assim, parece existir uma fonte não-neuronal de acetilcolina a nível do baço.
Um estudo recente mostrou que essas células serão provavelmente linfócitos t reguladores (que expressam recetores β2), sendo que a acetilcolina libertada poderá atuar em macrófagos esplénicos (via α7nAcCoR) induzindo um perfil anti-inflamatório.46-48 A Fig 2 esquematiza o funcionamento deste sistema.
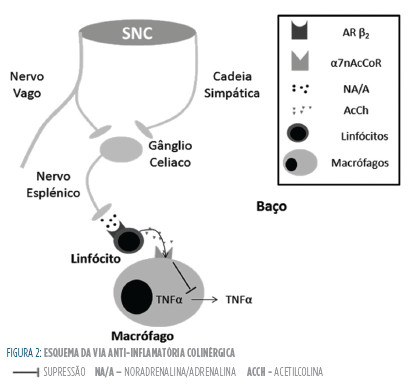
Considerando os pontos referidos, parece que a ativação do SNAPS induz um efeito predominante anti-inflamatório. Contudo, como foi dito relativamente ao SNAS, a supressão da resposta inflamatória na Sepsis pode ter efeitos dúbios: por um lado suprimindo o SIRS, mas por outro diminuindo a capacidade de eliminação bacteriana prolongando a resposta inflamatória a estes agentes. Na verdade, um estudo utilizando ratinhos knockout para o α7nAcCoR mostrou que estes possuíam uma maior capacidade de clearance bacteriano possivelmente devido a um maior recrutamento precoce de neutrófilos.49 Estes dados são coincidentes com o facto de o GtS-21 reduzir o recrutamento neutrofílico, embora independentemente da supressão do TNF-α.50
Impacto clínico
O SNA assume um papel de regulação de grande parte dos sistemas orgânicos incluindo o sistema imune. O conhecimento destes mecanismos de regulação pode permitir a criação de terapêuticas para patologias caracterizadas pela perturbação da resposta inflamatória/imune. Considerando que a Sepsis é um exemplo paradigmático deste tipo de patologias, a aplicação de terapêuticas moduladoras do SNA poderá trazer benefícios no melhor controlo da desregulação inflamatória.
A premissa de que a desregulação inflamatória tem um papel central na Sepsis levou à tentativa de abordar esta entidade com terapêuticas anti-inflamatórias. Contudo, o recurso a fármacos como o ibuprofeno,51 eritoran (antagonista do MD2/ TLR4),52 anticorpos monoclonais anti-TNF53 e antagonistas dos recetores do TNF e IL-154 foi marcado pelo insucesso. Por seu lado, os corticosteróides têm um papel ainda pouco definido na abordagem aos doentes sépticos, parecendo úteis apenas nos doentes com choque refratário à fluidoterapia e vasopressores.55 Considerando que a modulação do SNA produz efeitos predominantemente anti-inflamatórios, pode-se pensar que esta abordagem esteja condenada ao insucesso. Contudo, a modelação do SNA poderá abrir a oportunidade de controlar as respostas inflamatórias de forma mais fisiológica.
Por outro lado, o SNAS parece ter a dupla capacidade de ser anti e pró-inflamatório (Fig 3). Como já foi referido, este facto parece coincidir com a teoria da compartimentação da resposta imune. Contudo, pode ser especulado que esta capacidade permita ao SNAS modificar as respostas imunológicas consoante a necessidade do organismo, intensificando-a quando é necessário eliminar uma ameaça e suprimindo-aquando esta se torna deletéria.
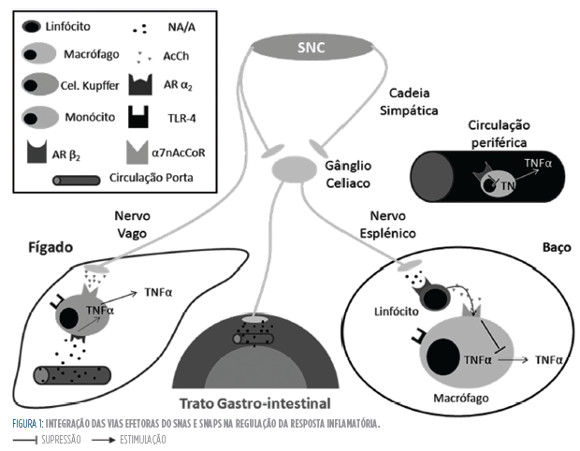
O papel que a estimulação simpática tem na estabilização dos doentes hemodinâmicos não pode ser esquecido. O recurso a aminas vasopressoras, como por exemplo a noradrenalina, é usado com o objetivo de assegurar a pressão de perfusão tecidual, apresentando um papel central no tratamento dos doentes sépticos. A hipoxia e a hipoperfusão são por si só fatores pró-inflamatórios pois ao induzirem isquemia, causam a libertação de substâncias pró-inflamatórias (damage-associated molecular patterns, DAMPs). Nesta perspetiva, as terapêuticas que otimizam a manutenção das condições de perfusão e oxIgEnação têm também um papel anti-inflamatório. Assim, as abordagens terapêuticas moduladoras do SNA, que serão explanadas a seguir, deverão respeitar necessidade de se manter uma estimulação a e β adrenérgicas necessárias à manutenção do débito cardíaco.
Considerando o que foi dito em relação ao SNAS, duas potenciais abordagens terapêuticas têm sido propostas: o recurso a agonistas dos recetores adrenérgicos β2 e antagonistas dos recetores α2.
1) Agonistas β2. O uso destes fármacos tem como objetivo reduzir a produção de citocinas pró-inflamatórias, minimizando o componente SIRS através da ativação dos recetores β2 nos macrófagos e linfócitos. De facto, esta abordagem aumentou a sobrevida em alguns modelos animais de Sepsis.46 Por outro lado, O recurso a β-bloqueadores não específicos, como o propranolol, Está associado a pior sobrevida em estudos animais,56 embora o mesmo não aconteça para os bloqueadores β1-selectivos.57 Curiosamente do entessépticos sobtratamento crónico com a β-bloqueadores parecem ter melhor sobrevida.58 Algumas condicionantes poderão limitar o uso deste tipo de abordagem. Por um lado, o efeito antiinflamatório, condicionando redução da produção de TNF-α, IL-1 e da função fagocitária, poderá causar maior dano do que benefício. Como já foi referido, o recurso a agentes biológicos anti-TNF-alfa e a recetores solúveis IL-1 foi tentado com insucesso em vários ensaios clínicos.35 Por outro lado, a estimulação de linfócitos com agonistas β2 induz um desvio da resposta imune para um perfil Th2 o que poderá implicar uma menor capacidade de resposta imune, comprometendo o clearance bacteriano. Por último, o recurso a agonistas β2 tem o potencial de gerar alterações hemodinâmicas deletérias para os doentes em choque, como por exemplo a indução de vasodilatação (por estimulação de recetores β2 do musculo liso das artérias) e o surgimento de arritmias.
2) Antagonistas α2. Esta abordagem baseia-se em estudos em modelo sanimais, que verificaram que a noradrenalina, libertada pelo trato gastrointestinal durante a Sepsis, estimula as células de Kupffer, induzindo a produção de citocinas pró-inflamatórias e podendo causar disfunção hepática.25 Portanto, o antagonismo de recetores α2, especificamente os α2A, poderia resultar numa menor ativação das células de Kupffere menor produção decitocinas pró inflamatórias e disfunção de órgão.24 Contudo, os recetores α2 estão também localizados pré-sinapticamente onde funcionam como um mecanismo de regulação da libertação de noradrenalina. Assim, em consequência do antagonismo deste recetor pode ocorrer um aumento da libertação de noradrenalina que atenue os efeitos anti-inflamatórios do antagonismo α2.
No que respeita ao SNAPS as abordagens que têm sido referidas são: Estimulação Nervosa vagal, agonistas α7nAcCoR, IACE e agonistas muscarínicos centrais.
3) Estimulação Nervosa vagal (VNS). A técnica de VNS por via transcutânea já é utilizada para o tratamento da depressão e epilepsia refratárias sendo que a sua aplicação em doentes sépticos já é tecnicamente possível. Em estudos animais a utilização de VNS permitiu reduzir os níveis séricos de citocinas precoces (IL-1 e TNFα) e tardias (HMGB1). A VNS não alterou de forma significativa os níveis séricos de IL-10. Para além disto, um estudo demonstrou melhoria da sobrevida no modelo de CLP (cecal ligation and puncture). Contudo, a VNS poderá induzir alterações hemodinâmicas, nomeadamente bradicardia, que seriam prejudicais no contexto da Sepsis. todavia, nos estudos animais o limiar de voltagem e frequência necessário para suprimir a resposta inflamatória é inferior ao necessário para reduzir a frequência cardíaca o que poderá contornar este problema.41
4) Agonistas α7nAcCoR. Vários agonistas foram testados (GTS-21, nicotina, colina, acetil-colina) verificando-se uma redução dos níveis séricos de HMGB1 e melhoria da sobrevida quando se utilizou GTS-21 ou nicotina no modelo CLP. A nicotina é um agonista inespecífico do α7nAcCoR, apresentando afinidade para muitos outros subtipos de recetores nicotínicos. Esta característica faz com que a nicotina possa induzir alterações hemodinâmicas nefastas para o doente séptico. O recurso a um agonista mais seletivo, como o GTS-21, poderá contornar estas dificuldades.59
5) Inibidores da ACE. Ao reduzirem a degradação da acetilcolina os IACE permitem prolongar a ação desta substância. No contexto da Sepsis, o objetivo passa por aumentar a quantidade de acetilcolina que se pode ligar ao α7nAcCoR a nível periférico e aos recetores m1 centrais. Num estudo com o modelo CLP, o recurso a IACE levou a redução da concentração plasmática de TNFα, IL-1β e il-6 (fisostigmina) assim como melhoria da sobrevida (neostigmina e fisostigmina). A grande limitação dos IACE prende-se com os efeitos hemodinâmicos podendo induzir bradicardia e hipotensão.41
Outro argumento a favor do uso das técnicas que estimulem o SNAPS advém da avaliação da variação espectral da frequência cardíaca e tensão arterial.60 Estudos recentes apontam que nos doentes sépticos há uma redução do espectro de variação da frequênciacardíacaassociadaaotónusdoSNAPS.61 Este facto apoia a ideia da existência de disfunção autonómica nos doentes sépticos que poderiam beneficiar da reversão desta disfunção com técnicas estimuladoras do SNAPS, como a estimulação vagal.7
Considerando o papel central que o uso de catecolaminas têm no paradigma atual do tratamento do choque séptico, o recurso a intervenções moduladoras do SNA com o objetivo de alterar a resposta imune só poderá ser posto em prática quando o papel deste sistema na patogénese da Sepsis for convenientemente esclarecido. Antes de se avançar com a aplicação clínica destas abordagens, algumas questões que deverão ser esclarecidas:
a) Qual a evolução temporal da atividade SNAS/ SNAPS ao longo da Sepsis?
b) A manipulação do SNAS/SNAPS implica instabilidade hemodinâmica? Se sim, esse risco é contrabalançado pelos benefícios inflamatórios/imunes?
c) O efeito anti-inflamatório destas abordagens induz redução significativa da capacidade de eliminação bacteriana?
d) Considerando que a Sepsis é um processo dinâmico qual o melhor timing para a aplicação destas terapêuticas?
Agradecimentos
Gostaria de agradecer ao meu orientador Professor Doutor António Sarmento pelo seu apoio e dedicação durante a realização deste trabalho. Gostaria também de reconhecer o seu contributo muito construtivo através da revisão crítica deste manuscrito.
Referências
1. Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein Am, Knaus WA, Schein Rm, Sibbald WJ. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCm Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care medicine. 1992. Chest 2009;136(5 Suppl):e28. [ Links ]
2. Angus DC, Linde-Zwirble Wt, Lidicker J, Clermont G, Carcillo J, Pinsky Mr. Epidemiology of severe sepsis in the united States: analysis of incidence, outcome, and associated costs of care. Crit Care med 2001; 29 (7): 1303-10. [ Links ]
3. Rittirsch D, Flierl MA, Ward PA. Harmful molecular mechanisms in sepsis. Nat rev immunol 2008;8(10):776-87. [ Links ]
4. Dellinger RP, Levy Mm, Carlet Jm, Bion J, Parker Mm, Jaeschke R, Reinhart K, Angus DC, Brun-Buisson C, Beale R, CalandraT, Dhainaut Jf, Gerlach H, Harvey M, Marini JJ, Marshall J, Ranieri M, Ramsay G, Sevransky J, Thompson Bt, Townsend S, Vender JS, Zimmerman Jl, Vincent Jl. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock: 2008. Crit Care med 2008;36(1):296-327. [ Links ]
5. Woiciechowsky C, Schoning B, Lanksch WR, Volk HD, Docke WD. Mechanisms of brain-mediated systemic anti-inflammatory syndrome causing immunodepression.[/ocontrib] J Mol Med (Berl) 1999;77(11):769-80.
6. Lyn-Kew K, Standiford TJ. Immunosuppression in sepsis. Curr Pharm Des 2008;14(19):1870-81. [ Links ]
7. Czura CJ, Tracey KJ. Autonomic neural regulation of immunity. J intern med 2005;257(2):156-66. [ Links ]
8. Barnes SJ,Ackland Gl. Beta-adreno receptor modulation of metabolic, end ocrine and immunologic function during criticalillness. Endocr metab immune Disord Drug targets2010;10(3):292-300. [ Links ]
9. Tracey KJ. The inflammatory reflex. Nature 2002;420(6917):85-9 [ Links ]
10. Munford RS, Tracey KJ. Is severe sepsis a neuroendocrine disease? Mol Med 2002;8(8):437-42. [ Links ]
11. Van Westerloo DJ. The vagal immune reflex: a blessing from above. Wien med Wochenschr 2010;160(5-6):112-7. [ Links ]
12. Akrout N, Sharshar T, Annane D. Mechanisms of brain signaling during sepsis. Curr Neuropharmacol 2009;7(4):296-301. [ Links ]
13. Hopkins SJ. Central nervous system recognition of peripheral inflammation: a neural, hormonal collaboration. Acta Biomed 2007;78 Suppl 1:231-47. [ Links ]
14. Zapata P, Larrain C, Reyes P, Fernandez R. Immunosensory signalling by carotid body chemoreceptors. Respir Physiol Neurobiol 2011;178(3):370-4. [ Links ]
15. Fernandez R, Nardocci G, Simon F, Martin A, Becerra A, Rodriguez-Tirado C, Maisey Kr, Acuna-Castillo C, Cortes PP. Lipopolysaccharide signaling in the carotid chemoreceptor pathway of rats with sepsis syndrome. Respir Physiol Neurobiol 2011;175(3):336-48. [ Links ]
16. Rudiger A, Stotz M, Singer M. Cellular processes in sepsis. Swiss Med Wkly 2008;138(43-44):629-34. [ Links ]
17. Liu Sf, Malik AB. NF - Kappa B activation as a pathological mechanism of septic shock and inflammation. Am J Physiol Lung Cell Mol Physiol 2006;290(4):l622-l45. [ Links ]
18. Elenkovi J,Wilderr l,Chrousos GP, vizi ES.Thesympatheticnerve--anintegrative interfacebetweentwosupersystems:thebrainandthe immunesystem. Pharmacol rev2000;52(4):595-638. [ Links ]
19. Bellinger DL, Millar BA, Perez S, Carter J, Wood C, Thyagarajan S, Molinaro C, Lubahn C, Lorton D. Sympathetic modulation of immunity: relevance to disease. Cell Immunol 2008; 252 (1-2): 27-56. [ Links ]
20. Szabo C, Hasko G, Zingarelli B, Nemeth ZH, Salzman Al, Kvetan V, Pastores SM, Vizi ES. Isoproterenol regulates tumour necrosis factor, interleukin-10, interleukin-6 and nitric oxide production and protects against the development of vascular hyporeactivity in endotoxaemia. Immunology 1997;90(1):95-100. [ Links ]
21. Grisanti lA, Evanson J, marchus E, Jorissen H, Woster AP, DeKrey W, Sauter Er, Combs CK, Porter JE. Pro-inflammatory responses in human monocytes are beta1-adrenergic receptor subtype dependent. mol immunol 2010;47(6):1244-54. [ Links ]
22. NakadatA,russellJA,BoydJH,Aguirre-Hernandezr,ThainKr,ThairSA,NakadaE,mcConechym,WalleyKr.beta2-Adrenergicreceptorgenepolymorphismisassociatedwithmortality in septic shock. Am J respir Crit Care med 2010;181(2):143-9. [ Links ]
23. Zhou m, Das P, Simms HH, Wang P. Gut-derived norepinephrine plays an important role in up-regulating IL-1beta and IL-10. Biochim Biophys Acta 2005;1740(3):446-52. [ Links ]
24. Zhang F, Wu R, Qiang X, Zhou M, Wang P. Antagonism of alpha 2A-adrenoceptor: a novel approach to inhibit inflammatory responses in sepsis. J Mol Med (Berl) 2010;88(3):289-96. [ Links ]
25. Miksa M, Wu R, Zhou M, Wang P. Sympathetic excitotoxicity in sepsis: pro-inflammatory priming of macrophages by norepinephrine. Front Biosci 2005;10:2217-29. [ Links ]
26. yang S, Koo DJ, Zhou m, Chaudry iH, Wang P. Gut-derived norepinephrine plays a critical role in producing hepatocellular dysfunction during early sepsis. Am J Physiol Gastrointest liver Physiol 2000;279(6):G1274-81. [ Links ]
27. yang S, Zhou m, Chaudry iH, Wang P. Norepinephrine-induced hepatocellular dysfunction in early sepsis is mediated by activation of alpha2-adrenoceptors. Am J Physiol Gastrointest liver Physiol 2001;281(4):G1014-21. [ Links ]
28. Cavaillon Jm, Annane D. Compartmentalization of the inflammatory response in sepsis and SIRS. J Endotoxin res 2006;12(3):151-70. [ Links ]
29. Kavelaars A, van de Pol m, Zijlstra J, Heijnen CJ. Beta 2-adrenergic activation enhances interleukin-8 production by human monocytes. J Neuroimmunol 1997;77(2):211-6. [ Links ]
30. Takahashi H, Kobayashi M, Tsuda Y, Herndon DN, Suzuki F. Contribution of the sympathetic nervous system on the burn-associate dimpairment of CCl3 production. Cytokine 2005; 29 (5): 208-14. [ Links ]
31. Kindt T GR, Osborne B. Kuby immunology 6th edition: WH freeman and Co; 2007. [ Links ]
32. manni m, Granstein rD, maestroni G. beta2-Adrenergic agonists bias TLR-2 and NOD2 activated dendritic cells towards inducing an IL-17 immune response. Cytokine 2011;55(3):380-6. [ Links ]
33. Deng J, Muthu K, Gamelli R, Shankar R, Jones SB. Adrenergic modulation of splenic macrophage cytokine release in polymicrobial sepsis. Am J Physiol Cell Physiol 2004;287(3):C730-6. [ Links ]
34. Suberville S, Bellocq A, fouqueray B, Philippe C, Lantz O, Perez J, Baud L. Regulation of interleukin-10 production by beta-adrenergic agonists. Eur J Immunol 1996;26(11):2601-5. [ Links ]
35. Sriskandan S, Altmann Dm. The immunology of sepsis. J Pathol 2008;214(2):211-23. [ Links ]
36. Freestone PP, Williams PH, Haigh RD, Maggs AF, Neal CP, Lyte M. Growth stimulation of intestinal commensal Escherichia coli by catecholamines: a possible contributory factor in trauma-induced sepsis. Shock 2002;18(5):465-70. [ Links ]
37. Straub RH, Pongratz G, Weidler C, Linde HJ, Kirschning CJ, Gluck T, Scholmerich J, Falk W. Ablation of the sympathetic nervous system decreases gram-negative and increases gram-positive bacterial dissemination: key roles for tumor necrosis factor/phagocytes and interleukin-4/lymphocytes. J Infect Dis 2005;192(4):560-72. [ Links ]
38. Link A, Selejan S, Maack C, Lenz M, Bohm M. Phosphodiesterase 4 inhibition but not beta-adrenergic stimulation suppresses tumor necrosis factor-alpha release in peripheral blood mononuclear cells in septic shock. Crit Care 2008;12(6):r159. [ Links ]
39. Pavlov VA, Ochani M, Gallowitsch-Puerta M, Ochani K, Huston JM, Czura CJ, Al-Abed Y, Tracey KJ. Central muscarinic cholinergic regulation of the systemic inflammatory response during endotoxemia. Proc Natl Acad Sci USA 2006;103(13):5219-23. [ Links ]
40. Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Yang H, Ulloa L, Al-Abed Y, Czura CJ, Tracey KJ. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003;421(6921):384-8. [ Links ]
41. Rosas-Ballina M, Tracey KJ. Cholinergic control of inflammation. J intern med 2009;265(6):663-79. [ Links ]
42. Pavlov VA, Parrish WR, Rosas-Ballina M, Ochani M, Puerta M, Ochani K, Chavan S, Al-Abed Y, Tracey KJ. Brain acetylcholinesterase activity controls systemic cytokine levels through the cholinergic anti-inflammatory pathway. Brain Behav Immun 2009;23(1):41-5. [ Links ]
43. Huston JM, Ochani M, Rosas-Ballina M, Liao H, Ochani K, Pavlov VA, Gallowitsch-Puerta M, Ashok M, Czura CJ, Foxwell B, Tracey KJ, Ulloa L. Splenectomy inactivates the cholinergic antiinflammatory pathway during lethal endotoxemia and polymicrobial sepsis. J Exp Med 2006;203(7):1623-8. [ Links ]
44. Rosas-Ballina M, Ochani M, Parrish WR, Ochani K, Harris YT, Huston JM, Chavan S, Tracey KJ. Splenic nerve is required for cholinergic antiinflammatory pathway control of TNF in endotoxemia. Proc Natl Acad Sci USA 2008;105(31):11008-13. [ Links ]
45. Vida G, Pena G, Deitch EA, Ulloa L. Alpha7-cholinergic receptor mediates vagal induction of splenic norepinephrine. J Immunol 2011;186(7):4340-6. [ Links ]
46. Vida G, Pena G, Kanashiro A, Thompson-Bonilla Mdel R, Palange D, Deitch EA, Ulloa L. Beta2-Adrenoreceptors of regulatory lymphocytes are essential for vagal neuromodulation of the innate immune system. Faseb J 2011; 25 (12): 4476-85. [ Links ]
47. Rosas-Ballina M, Olofsson PS, Ochani M, Valdes-Ferrer SI, Levine YA, Reardon C, Tusche MW, Pavlov VA, Andersson U, Chavan S, Mak TW, Tracey KJ. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science 2011;334(6052):98-101. [ Links ]
48. Pena G, Cai B, Ramos L, Vida G, Deitch EA, Ulloa L. Cholinergic regulatory lymphocytes re-establish neuromodulation of innate immune responses in sepsis. J Immunol 2011;187(2):718-25. [ Links ]
49. Giebeleni A, Van Westerloo DJ, Larosa GJ, Devos AF, Van Der Poll T. Stimulation of alpha 7 cholinergic receptors inhibits lipopolysaccharide-induced neutrophil recruitment by a tumor necrosis factor alpha-independent mechanism. Shock 2007;27(4):443-7. [ Links ]
50. SaeedrW, varmaS, Peng-Nemeroff t, Sherry B, Balakhaneh D, HustonJ, tracey KJ,Al-Abed y, metzCN. Cholinergicstimulationblocks endothelial cell activation andleukocyte recruitment during inflammation. J Exp med 2005;201(7):1113-23. [ Links ]
51. Bernard Gr, Wheeler AP, russell JA, Schein r, Summer Wr, Steinberg KP, fulkerson WJ, Wright PE, Christman BW, Dupont WD, Higgins SB, Swindell BB. The effects of ibuprofen on the physiology and survival of patients with sepsis. The ibuprofen in Sepsis Study Group. N Engl J med 1997;336(13):912-8. [ Links ]
52. Opal SM, Laterre PF, Francois B, Larosa SP, Angus DC, Mira JP, Wittebole X, Dugernier T, Perrotin D, Tidswell M, Jauregui L, Krell K, Pachl J, Takahashi T, Peckelsen C, Cordasco E, Chang CS, Oeyen S, Aikawa N, Maruyama T, Schein R, Kalil AC, Van Nuffelen M, Lynn M, Rossignol DP, Gogate J, Roberts MB, Wheeler JL, Vincent JL, Group AS. Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: the ACCESS randomized trial. JAMA 2013;309(11):1154-62. [ Links ]
53. Abraham E, Wunderink R, Silverman H, Perl TM, Nasraway S, Levy H, Bone R, Wenzel RP, Balk R, Allred R, et al. Efficacy and safety of monoclonal antibody to human tumor necrosis factor alpha in patients with sepsis syndrome. A randomized, controlled, double-blind, multicenter clinical trial. TNF-alpha MAb Sepsis Study Group. JAMA 1995;273(12):934-41. [ Links ]
54. Opal Sm, fisher CJ, Jr., Dhainaut Jf, vincent Jl, Brase r, lowry Sf, Sadoff JC, Slotman GJ, levy H, Balk rA, Shelly mP, Pribble JP, laBrecque Jf, lookabaugh J, Donovan H, Dubin H, Baughman r, Norman J, Demaria E, matzel K, Abraham E, Seneff m. Confirmatory interleukin-1 receptor antagonist trial in severe sepsis: a phase iii, randomized, double-blind, placebo-controlled, multicenter trial. The interleukin-1 receptor Antagonist Sepsis investigator Group. Crit Care med 1997;25(7):1115-24. [ Links ]
55. Minneci PC, Deans KJ, Natanson C. Corticosteroid therapy for severe sepsis and septic shock. JAMA 2009;302(15):1643; author reply 4-5. [ Links ]
56. Lang CH, Nystrom G, Frostr A. Beta-adrenergic blockade exacerbates sepsis-induced changes in tumor necrosis factor alpha and interleukin- 6 in skeletal muscle and is associated with impaired translation initiation. J Trauma 2008;64(2):477-86. [ Links ]
57. Mori K, Morisaki H, Yajima S, Suzuki T, Ishikawa A, Nakamura N, Innami Y, Takeda J. Beta-1 blocker improves survival of septic rats through preservation of gut barrier function. Intensive Care Med 2011;37(11):1849-56. [ Links ]
58. Macchia A, Romero M, Comignani PD, Mariani J, DEttorre A, Prini N, Santopinto M, Tognoni G. Previous prescription of beta-blockers is associated with reduced mortality among patients hospitalized in intensive care units for sepsis. Crit Care Med 2012;40(10):2768-72. [ Links ]
59. tracey KJ. reflex control of immunity. Nat rev immunol 2009;9(6):418-28. [ Links ]
60. Tang CH, Chan GS, Middleton PM, Savkin AV, Lovell NH. Spectral analysis of heart period and pulse transit time derived from electrocardiogram and photoplethysmogramin sepsis patients. Conf Proc IEEE Eng Med Biol Soc 2009;2009:1781-4. [ Links ]
61. Pancoto JA, Correa PB, Oliveira-Pelegrin GR, Rocha MJ. Autonomic dysfunction in experimental sepsis induced by cecal ligation and puncture. Auton Neurosci 2008;138(1-2):57-63. [ Links ]
Paulo Andrade
Faculdade de Medicina, universidade do Porto. 4200-319, Porto. E-mail: joao_c_goncalves@hotmail.com
Data de recepção / reception date: 20/05/2013
Data de aprovação / approval date: 26/07/2013

