










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista Portuguesa de Imunoalergologia
versão impressa ISSN 0871-9721
Rev Port Imunoalergologia vol.23 no.3 Lisboa set. 2015
CASO CLÍNICO
Angioedema hereditário com complemento normal: A importância do estudo genético na família
Hereditary angioedema with normal C1 inhibitor: The importance of a family genetic study
Joana Cosme1; Amélia Spínola Santos1; Anabela Lopes1; António Martinho2; Manuel Pereira Barbosa1,3
1 Centro Hospitalar Lisboa Norte – Hospital de Santa Maria, Lisboa
2 Instituto Português do Sangue e da Transplantação – Centro Português do Sangue e da Transplantação de Coimbra, Coimbra
3 Universidade de Lisboa – Faculdade de Medicina, Lisboa
RESUMO
O angioedema hereditário (AEH) com complemento normal não se associa a alterações quantitativas ou funcionais do C1 inibidor. Cerca de 25% dos europeus com angioedema hereditário (AEH) com complemento normal apresentam mutação do gene F12 que codifica o fator XII da coagulação e, em alguns doentes, condições associadas a níveis elevados de estrogénios como gravidez, contracetivos orais e terapêutica hormonal de substituição são desencadeantes de crise. Neste artigo descreve‑se o caso de uma doente, tal como a sua mãe, com o diagnóstico de AEH com complemento normal e identificação de mutação c.1032 C> A no gene F12, com crises de angioedema recorrentes, por vezes associadas a condições com elevados níveis de estrogénios (contracetivos orais, anel vaginal e nas duas gravidezes). O estudo genético das suas descendentes permitiu identificar uma mutação missense c.1032C> A e uma mutação frameshift (ainda não descrita) na segunda descendente. Este caso demonstra que a pesquisa de mutações no gene do F12 permite, em descendentes de doentes com esta patologia, estabelecer um diagnóstico precoce e evitar a exposição a fatores desencadeantes de crise.
Palavras‑chave: Angioedema hereditário com complemento normal, fator XII, gene F12, mutação frameshift, mutação missense.
ABSTRACT
Hereditary angioedema (HAE) with normal C1 inhibitor (C1 INH) is associated with normal levels and activity of C1INH. Mutations in F12 gene that codifies for coagulation factor XII are reported to occur in up to 25% of Europeans with this condition. In some patients high‑leveled estrogenic conditions such as oral contraceptives, hormone substitution therapy and pregnancy exacerbate the disease. The authors describe a case of a female that, like her mother, has the diagnosis of HAE with normal C1INH with a missense mutation c.1032 C>a on F12 gene, with history of recurrent episodes of oedema sometimes associated with high‑leveled estrogenic conditions (oral contraceptives, vaginal ring and pregnancies). Genetic study of patients offspring found a missense mutation c.1302 C>A and a frameshift mutation on F12 gene (not described yet) on the youngest daughter. The aim of this article is to show that searches for F12 mutations in offspring of patients with HAE with normal C1INH allows an early diagnosis and suspension of the triggering factors.
Key‑words: F12 gene, factor XII, frameshift mutation, hereditary angioedema with normal C1 inhibitor, missense mutation.
INTRODUÇÃO
O angioedema hereditário (AEH) é uma doença genética rara que se carateriza por episódios recorrentes de edema não pruriginoso localizado ao tecido subcutâneo ou submucoso e que pode estar associado a risco de vida, nomeadamente quando existe envolvimento das vias aéreas1,2.
Consensos recentes defendem que o AEH deve ser classificado em duas subcategorias: com base nos níveis e atividade do C1INH: 1‑AEH com complemento normal (normal C1‑INH); 2‑AEH com défice de C1INH. Sendo esta última categoria subdividida em défice quantitativo (AEH tipo 1) ou qualitativo (AEH tipo 2) de C1INH3.
A primeira descrição de AEH com C1 INH normal data de 1986 por Warin et al.4. Uma das primeiras séries de doentes é apresentada, cerca de 14 anos mais tarde, em 2000, por Bork K et al.5, e as bases moleculares do AEH com C1 INH normal são descritas em 2006 por Dewald e Bork6 e por Cichon et al.7.
Várias têm sido, ao longo do tempo, as designações para esta patologia: novo AEH, AEH tipo 3, AEH estrogenio‑dependente3. Atualmente, defende‑se a denominação de AEH com C1INH normal, já que o termo tipo 3 não permite uma distinção clara entre este e o AEH tipos 1 e 2, nem é suficiente classificar os diferentes subtipos de doentes com AEH com C1INH normal. Para além disto, a designação estrogenio‑dependente não deve ser utilizada porque, apesar dos estrogénios poderem ser um fator desencadeante de crises em muitos doentes, tal não se verifica em todos os doentes com AEH com C1INH normal3.
A maior coorte de doentes com AEH com C1INH normal foi relatada por Bork et al. que, em 2009, descreveu 53 famílias com esta patologia8. Do ponto de vista clínico, o AEH com C1INH normal carateriza‑se, à semelhança das formas de AEH com défice de C1INH, por episódios de angioedema recorrentes sem urticária associada.
O edema pode atingir qualquer órgão ou sistema havendo contudo um envolvimento da face, língua e vias aéreas, é mais frequentemente descrito do que nos casos de AEH com défice de C1INH. Por esta razão, o risco de asfixia fatal não deve ser ignorado9,10.
Os mecanismos fisiopatológicos que estão na base do AEH com C1INH normal não estão ainda totalmente esclarecidos, embora se pense que a bradicinina seja o mediador envolvido neste processo3.
Trata‑se de uma patologia autossómica dominante com penetrância incompleta1,3 em que mutações no gene F12 que codifica o fator XII da coagulação (fator de Ha‑geman), isto é, um fator cuja ativação desencadeia a cascata produtora de cininas, parecem estar na base das alterações clinico‑patologicas que ocorrem no AEH com C1INH normal6. Em 2006 foram identificadas duas mutações missense diferentes associadas ao AEH com C1INH normal. Estas localizam‑se ambas no exão 9 e no mesmo locus (5q33‑qter) do gene F12 (Online Mendelian Inheritance in Man #610619), sendo que ambas se traduzem numa substituição aminoacídica – a mutação c.1032C> A que origina uma substituição do aminoácido treonina por lisina (Thr309Lys) e a mutação c.1032C> G que leva a uma substituição do aminoácido treonina por arginina (Thr309Arg) 6,7,10,11. Não foram encontradas diferenças nos doentes com as mutações Thr309Lys e Thr309Arg3; no entanto, enquanto a última mutação é pouco frequente, a primeira surge em cerca de 25% dos europeus com AEH com C1INH normal3,6. Ambas as mutações parecem ser raras na população americana3. Uma outra mutação no gene F12 foi recentemente encontrada numa única família turca com AEH com C1INH normal (deleção 72‑bp – c.971_1018; 24del72) 11,13.
CASO CLÍNICOMulher de 40 anos, natural de Espanha, residente em Faro, com antecedentes de hipotiroidismo, com uma primeira crise de angioedema facial aos 18 anos, após a realização de um procedimento estomatológico invasivo.
Aos 27 anos, na sequência da toma de contracetivos orais (CO), apresenta várias crises recorrentes de AE da face e membros que conduziram à suspensão dos CO cerca de três meses mais tarde, com resolução das crises.
Aos 30 anos tem uma nova crise de AE facial com urticária associada à toma de metamizol prescrito por odinofagia. Nesta data, ainda a residir em Espanha, e‑lhe feito o diagnóstico de hipersensibilidade ao metamizol (tolera ibuprofeno e paracetamol).
Aos 35 anos, após colocação de anel vaginal, reinicia crises de AE frequentes, com envolvimento facial e abdominal, pelo que recorre várias vezes à urgência. Na investigação das crises abdominais fez endoscopia alta e baixa, num único tempo, e que se complicou de AE facial.
O diagnóstico de AEH com C1 INH normal foi feito em Espanha quando a doente se encontrava grávida (1.º trimestre da 1.ª gravidez). Foi feito estudo seriado do complemento, nomeadamente com doseamento de C1 inibidor e avaliação da função C1 inibidor, que se encontravam os sempre dentro dos limites da normalidade e, ainda, pesquisa de mutações do gene F12 com identificação de uma mutação c.1032C> A (Thr328Lys). Esta gravidez decorreu com crises abdominais e faciais. Concomitantemente, foi feito o estudo genético da mãe da doente que, aparentemente, tinha história de angioedema facial recorrente nunca investigado e que confirmou a existência de uma mutação genética idêntica à da filha.
Aos 39 anos, já em Portugal (Faro), no 1.º trimestre da sua segunda gravidez, a doente desenvolve uma crise de AE facial exuberante com necessidade de terapêutica com plasma fresco congelado. Foi efetuado plasma fresco, uma vez que o Hospital de Faro dispunha apenas de icatibant, fármaco que não é recomendado nem na gravidez nem em crianças. Por controlo clínico incompleto foi contatado o Hospital de Santa Maria que providenciou o envio de concentrado de C1 inibidor para administração.
O parto foi eutócico sem intercorrências, tendo a doente feito concentrado de C1 inibidor antes do parto.
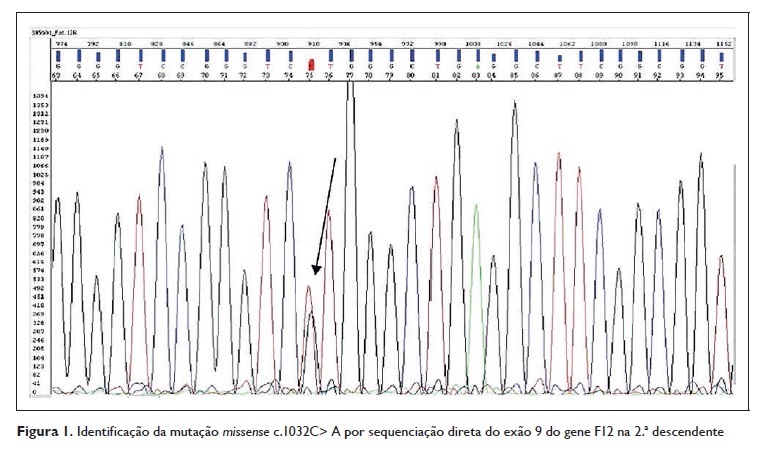
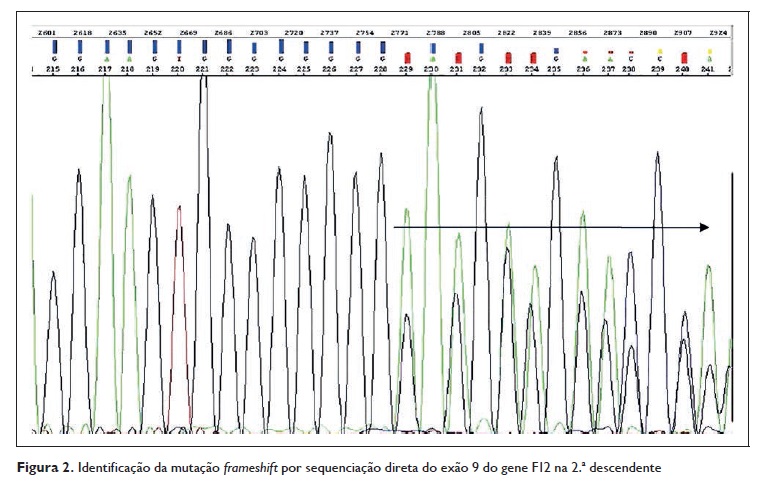
Em 2014 realizou‑se, no Centro do Sangue e da Transplantação de Coimbra, o estudo genético das duas filhas da doente, ambas do sexo feminino. Após sequenciação direta do exão 9 do gene F12, foram encontradas duas mutações na 2.ª descendente: uma mutação missense c.1302C> A (Thr328Lys) igual à da sua mãe e avó e uma mutação frameshift no gene F12 ainda não descrita e cujas consequências a nível dos transcritos alternativos do gene F12 não são ainda conhecidas (Figuras 1 e 2).
A doente tem dois irmãos, homens, sem clínica de angioedema. As sobrinhas da doente, filhas dum dos irmãos da doente, e sem clínica de angioedema, aguardam realização de estudo genético em Espanha.
DISCUSSÃO
O AEH com C1INH normal apresenta clínica semelhante ao AEH tipos 1 e 2 apesar de laboratorialmente se caracterizar por níveis normais de concentrado de C1 inibidor.
Deve‑se suspeitar desta patologia em doentes com angioedema recorrente sem urticária associada, com história familiar de angioedema recorrente sem fator desencadeante identificado ou associado a condições associadas a elevados níveis de estrogénios (gravidez, contracetivos orais, terapêutica hormonal de substituição). Mutações no gene F12 são as únicas mutações conhecidas, até ao momento, associadas a esta patologia e que, apesar de estarem presentes apenas numa minoria dos doentes, confirmam o diagnóstico.
O caso clínico descrito é ilustrativo duma família onde se encontra uma mutação missense c.1032C> A no gene do F12. Atualmente, defende‑se que devem ser pesquisadas mutações no gene F12 em indivíduos pertencentes a famílias onde existem mais do que dois doentes com o diagnóstico de AEH com C1INH normal3,8. Com base neste pressuposto e, tendo em conta que na família descrita no caso clínico apresentado existiam dois elementos com o diagnóstico de AEH com C1INH normal e confirmação genética da existência de mutação c.1032 C> A (a doente e a sua mãe), procedeu‑se à pesquisa de mutações no gene F12 nas duas descendentes. Bork et al. (2013) 13 defende que é importante confirmar-se o diagnóstico de AEH com C1INH normal por três razões: a primeira porque esta patologia pode associar‑se a crises de angioedema potencialmente fatais (risco de asfixia); a segunda porque o AEH com mutação no gene F12 é uma doença autossómica dominante e, como tal, a mutação pode ser transmitida aos descendentes; e, a terceira, porque a mutação pode estar presente em membros não afetados de famílias de doentes com AEH com C1INH normal e que, como tal, estão em risco de desenvolver crises de AE na presença de determinados fatores desencadeantes de crise, como por exemplo CO, terapêutica hormonal de substituição, gravidez e toma de inibidores da enzima conversão da angiotensina (IECA).
Assim, nesta família, a pesquisa de mutações no gene F12 nas descendentes permite não só identificar o diagnóstico a tempo de desenvolver medidas terapêuticas preventivas adequadas, nomeadamente profilaxia de crise de angioedema nos procedimentos medico‑cirurgicos, como também identificar a necessidade de evicção de terapêutica estrogénica e IECA.
Outro aspeto importante neste caso clínico consiste na identificação de uma mutação frameshift no gene F12 que até ao momento ainda não foi descrita e cujas repercussões, a nível da doença, não são conhecidas. Assim, apesar desta mutação não se traduzir em termos proteicos, as suas repercussões na doença são ainda desconhecidas, podendo este trabalho ser o ponto de partida para estudos futuros.
CONCLUSÃO
A pesquisa de mutações do gene F12 em familiares de doentes com AEH com C1INH normal com mutação identificada é importante não só para estabelecimento de um diagnóstico como também para a adoção de medidas preventivas e terapêuticas adequadas.
REFERÊNCIAS
1. Walford HH, Zuraw BL. Current update on cellular and molecular mechanisms of hereditary angioedema. Ann Allergy Asthma Immunol 2014; 112: 413‑8. [ Links ]
2. Betschel SBadiou J, Binkley K, Hébert J, Kanani A, Keith P et al. Canadian hereditary angioedema guideline. Allergy Asthma Clin Immunol 2014; 10:50. [ Links ]
3. Zuraw BL, Bork K, Binkley KE, Banerji A, Christiansen SC, Castaldo A, et al. Hereditary angioedema with normal C1 inhibitor function: Consensus of an international expert panel. Allergy Asthma Proc 2012; 33 (Suppl 1):S145‑56. [ Links ]
4. Warin RP, Cunliffe WJ, Greaves MW, Wallington TB. Recurrent angioedema: familial and oestrogen‑induced. Br J Dermatol 1986; 115:731‑4. [ Links ]
5. Bork K, Barnstedt SE, Koch P, Traupe H. Hereditary angioedema with normal C1‑inhibitor activity in women. Lancet 2000; 356:213‑7. [ Links ]
6. Dewald G, Bork K. Missense mutations in the coagulation factor XII (Hageman factor) gene in hereditary angioedema with normal C1 inhibitor. Biochem Biophys Res Commun 2006; 343:1286‑9. [ Links ]
7. Cichon S, Martin L, Hennies HC, Müller F, Van Driessche K, Karpushova A, et al. Increased activity of coagulation factor XII (Hageman Factor) causes hereditary angioedema type III. Am J Hum Genet 2006; 79: 1098‑104. [ Links ]
8. Bork K, Wulff K, Hardt J, Witzke G, Staubach P. Hereditary angioedema caused by missense mutations in the factor XII gene: clinical features, trigger factors, and therapy. J Allergy Clin Immunol 2009;124:129‑34. [ Links ]
9. Riedl, M. Hereditary angioedema with normal C1 INH (HAE type III). J Allergy Clin Immunol Pract 2013;1:427‑32. [ Links ]
10. Bork K. Diagnosis and treatment of hereditary angioedema with normal C2 inhibitor. Allergy Asthma Clin Immunol 2010; 6:1‑18. [ Links ]
11. Bork K, Wulff K, Meinke P, Wagner N, Hardt J, Witzke G. A novel mutation in the coagulation factor 12 gene in subjects with hereditary angioedema and normal C1‑inhibitor. Clin Immunol 2011;141:31‑5. [ Links ]
12. Marcos C, López Lera A, Varela S, Liñares T, Alvarez‑Eire MG, Lopez‑Trascasa M. Clinical, biochemical and genetic characterization of type III hereditary angioedema in 13 Northwest Spanish families. Ann Allergy Asthma Immunol 2012; 109:195‑200. [ Links ]
13. Bork K, Wulff K, Gunther W, Stanger C, Lohse P, Hardt J. Antihistamine‑resistant angioedema in women with negative family history: Estrogens and F12 gene mutations. Am J Med 2013;126:1142.e9‑14 [ Links ]
Joana Cosme
Serviço de Imunoalergologia do Hospital de Santa Maria,
Centro Hospitalar Lisboa Norte
Avenida Professor Egas Moniz
1649‑035 Lisboa
E‑mail: joanamcosme@gmail.com
Financiamento: Nenhum.
Declaração de conflitos de interesse: Nenhum.
Data de receção / Received in: 05/02/2015
Data de aceitação / Accepted for publication in: 28/05/2015


{kind=link}
{kind=link}