










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
versão impressa ISSN 0872-0169
Port J Nephrol Hypert vol.27 no.3 Lisboa set. 2013
Different pathways and biomarkers of acute and chronic cyclosporineinduced nephrotoxicity in a rat model – focus on overexpression of mTOR and Mki67
Diferentes vias de sinalização e biomarcadores da nefrotoxicidade aguda e crónica induzida pela Ciclosporina num modelo de rato – enfoque para a sobreexpressão de mTOR e Mki67
Jose Sereno1, Paulo Rodrigues-Santos2,3, Helena Vala4,5, Belmiro Parada6, Rui Alves7,8, Edite Teixeira-Lemos4,5, Petronila Rocha-Pereira9, Frederico Teixeira1, Flavio Reis1*
1Laboratory of Pharmacology & Experimental Therapeutics, IBILI, Faculty of Medicine, University of Coimbra, Portugal
2Institute of Immunology, Faculty of Medicine, University of Coimbra, Portugal
3Immunology and Oncology Laboratory, Centre for Neurosciences and Cell Biology, Coimbra, Portugal
4,5ESAV and Educational, Technologies and Health Study Centre, Polytechnic Institute of Viseu, Portugal
6,7Department of Urology and Renal Transplant and Department of Nephrology, CHUC, Coimbra, Portugal
8University Nephrology Unit, Faculty of Medicine, University of Coimbra, Portugal
9Research Centre for Health Sciences, Beira Interior University, Covilhã,Portugal.
ABSTRACT
Calcineurin inhibitors, in particular Cyclosporin A (CsA), remains the cornerstones of immunosuppressive regimens in many transplantation centres worldwide, regardless of drug-induced nephrotoxicity. The pathogenesis of CsA-induced nephropathy remains to be fully elucidated, but seems to be affected by the duration of drug exposure. This study aimed to clarify the molecular pathways involved in acute and chronic CsA-induced nephrotoxicity, focusing on serum, urinary and renal markers. The study comprised 24 male Wistar rats, divided in two models: acute and chronic CsA (5 mg/Kg bw/day) exposure (3 vs 9 weeks) vs matched control groups. The following data was evaluated: blood pressure and heart rate; serum total and non-HDL cholesterol, glucose and insulin; serum and urine creatinine, blood urea nitrogen (BUN), clearances and glomerular filtration rate (GFR); serum, urine and kidney tissue lipid peroxidation, via malondialdehyde (MDA); kidney mRNA expression of proliferative markers (PCNA, TGF-β1, mTOR and Mki67); kidney lesions. CsA has promoted hypertension and tachycardia, which were aggravated with the duration of exposure. Creatinine and BUN clearance and GFR showed early renal dysfunction, accompanied by increase serum creatinine (p<0.05) and BUN (p<0.01) levels, as well as kidney lipid peroxidation (p<0.05), which worsened with chronic exposure. Renal lesions were evident only after the chronic treatment. However, acute CsA exposure induced PCNA and TGF- β1 kidney mRNA up-regulation (p<0.05), unchanged mTOR and down-regulation of Mki67, while chronic treatment revealed a normalized PCNA and TGF- β 1 expression, accompanied by prominent mTOR and Mki67 up-regulation (p<0.01). In conclusion, CsA-induced nephrotoxicity is aggravated over time and distinct mechanisms and markers are involved in acute and chronic exposure. Chronic nephrotoxicity is accompanied with intense overexpression of mTOR and Mki67. These findings reinforce the rationale for early substitution of CsA by less nephrotoxic agents, being mTOR inhibitors a validated choice, in order to prevent chronic CsAinduced nephrotoxicity.
Key-words: acute and chronic cyclosporine-induced nephrotoxicity; biomarkers; gene expression; histology; rat model.
RESUMO
Introdução: Os inibidores da Calcineurina, em particular a Ciclosporina (CsA), continua a ser um dos pilares da terapêutica imunossupressora em muitos centros de transplantação por todo o mundo, não obstante a nefrotoxicidade a eles associada. A patogénese da nefropatia induzida pela CsA ainda permanece por elucidar completamente, mas sabe-se que e afetada pelo tempo de exposição ao fármaco. Este estudo teve como objetivo esclarecer as vias moleculares envolvidas na nefrotoxicidade aguda e cronica induzidas pela CsA, com especial enfoque em marcadores séricos, urinários e renais. Grupos e métodos: O estudo envolveu 24 ratos Wistar machos, divididos em dois modelos: exposição aguda e cronica (3 vs 9 semanas) a CsA (5 mg/kg de peso corporal/dia) versus respectivos grupos controlo. Foram avaliados os seguintes parâmetros: pressão arterial e frequência cardíaca; colesterol total e colesterol não-HDL, glicose e insulina sericas, creatinina e azoto ureico em soro e urina, clearances e taxa de filtração glomerular (GFR), peroxidação lipídica em soro, urina e rim (medição de malondialdeido); quantificação do mRNA renal de marcadores de proliferação (PCNA, TGF-β1, mTOR e Mki67); avaliação histomorfologica. Resultados: A CsA promoveu hipertensão e taquicardia, que foram agravadas com a duração da exposição. Os valores das clearance de creatinina e azoto ureico e da GFR mostraram disfunção renal precoce, acompanhada por um aumento dos níveis de creatinina serica ( p<0,05) e azoto ureico (p<0,01), assim como de peroxidação lipídica no rim (p<0,05), que se agravaram com a exposição cronica. As lesões renais foram evidentes somente apos o tratamento cronico. Contudo, a exposição aguda a CsA induziu a sobreexpressão do mRNA de PCNA e de TGF- β1 a nível renal (p <0,05), sem alteração de mTOR e com redução da expressão do Mki67. O tratamento cronico revelou níveis normalizados de mRNA de PCNA e de TGF-β1, acompanhados por uma notória sobreexpressão renal de mTOR e de Mki67 (p< 0,01). Conclusão: A nefrotoxicidade induzida por ciclosporina e agravada com o tempo e existem mecanismos e marcadores distintos para a exposição aguda e cronica a CsA. A nefrotoxicidade cronica e acompanhada de intensa sobreexpressão de mTOR e de Mki67. Estes resultados reforçam a necessidade da substituição precoce de CsA por agentes menos nefrotoxicos, podendo os inibidores da mTOR ser uma escolha valida, a fim de prevenir a nefrotoxicidade cronica induzida pela ciclosporina.
Palavras-chave: biomarcadores; expressão genica; histologia; nefrotoxicidade aguda e cronica induzida pela ciclosporina; rato.
INTRODUCTION
Cyclosporine A (CsA), a calcineurin inhibitor (CNI), remains a pivotal immunosuppressive drug to prevent allograft rejection. Its introduction led to a significant reduction in the incidence of acute rejection and an improvement in transplant survival1,2. However, the clinical use of CsA is often limited by severe sideeffects, including drug-induced hypertension and nephrotoxicity3-5. Renal dysfunction is an independent risk factor for graft loss and mortality after kidney transplantation (KTx) and cardiovascular disease (CVD) is the main cause of death post-KTx6-8; thus, extended long-term graft survival has not been completely achieved.
The recognition of these serious adverse effects sparked interest in CsA-sparing strategies9: dose reduction is associated with a modest improvement in renal function, but CsA-induced nephropathy is progressive over time when exposure is maintained; CsA avoidance is associated with high acute rejection rates and is not an option; minimization protocols are the current preferred therapy, including the conversion from CsA to other drugs, especially to Sirolimus (SRL), an inhibitor of the mammalian target of rapamycin10,11.
Late conversion from CsA to SRL has achieved variable results, possibly because withdrawal was attempted after the kidney damage was already too extensive. Early conversion, on the other hand, prior to significant graft damage, has generally improved creatinine clearance and markers of fibrosis, and decreased chronic allograft lesions, but the issue is far from consensual, as the Convert trial has demonstrated12-15. Complete avoidance of CNIs, in particular of CsA, from transplantation immunotherapy, has been viewed as an invalid option by almost all the transplantation centres worldwide, particularly because of the risks in acute rejection. The major question nowadays concerning the protocols of immunotherapy is to find the most adequate duration for CsA exposure and the proper moment for replacement by other less nephrotoxic drugs, without compromising the graft by a rejection episode. However, the knowledge of these aspects imposes better clarification of the histomorphological and molecular mechanisms associated with acute versus chronic exposure, which is largely dependent on experimental studies.
Previous studies from our group, as well as others, have already elucidated, in KTx patients, as well as in animal models, some of the molecular events involved in the cardiovascular side-effects of CsA, including vascular hyperreactivity, NO impairment, oxidative stress and sympathetic nervous system overactivity16-19. Despite the efforts to identify mechanisms of CsA-induced renal toxicity20,21, whether by acute or chronic exposure, much still remains to be elucidate.
New efforts to try and identify molecular mechanisms, as well as important markers in CsA-induced early renal toxicity will greatly help improve clinical practice choices concerning immunosuppressive regimens.
Drug safety evaluation has mainly been based on biochemical and histopathological data, but transcriptional profiling has the promise of being able to detect toxicity objectively, and the gene expression changes associated with toxicity may also assist with our understanding of the mechanism of certain drug induced accurately and earlier toxicity, and have been increasingly used clinically and analytically22,23.
Early diagnosis of nephropathy can greatly improve patient diagnosis, but the initial stages of CsAinduced nephropathy are largely asymptomatic, making early diagnosis difficult20. Since the current diagnostic techniques employed to detect CsA nephropathy seem to be unsatisfactory, the identification of novel, early disease indicators is currently a major research focus. Several emergent biomarkers putatively involved in drug-induced nephrotoxicty have been evaluated during the last years, including kidney injury molecule-1 (KIM-1) and neutrophil gelatinaseassociated lipocalin (NGAL), among others. In this study, we intended to elucidate some of the important pathways and putative biomarkers involved in CsA-induced acute and chronic nephrotoxicity in an animal model, focusing on important mediators of proliferation and fibrosis, which are key pathways for aggravation of nephropathy.
RESEARCH DESIGN AND METHODS
Animals and treatments
Twenty four male Wistar rats (Charles River Lab. Inc, Barcelona, Spain), eleven weeks of age, were maintained in an air conditioned room, subjected to 12h dark/light cycles and given standard laboratory rat chow (IPM-R20, Letica, Barcelona, Spain) and free access to tap water. The animals were divided into two groups: acute and chronic CsA (5 mg/Kg/day of Sandimun NeoralR) exposure, of 3 and 9 weeks respectively, versus the corresponding control groups (vehicle), with an n=6 per group. Treatments were performed by oral gavage. Body weight was monitored throughout the treatment period. Animal experiments were conducted according to the European Council Directives on Animal Care and to the National Authorities. Cyclosporine (Sandimun Neoral R) was obtained from Novartis Farma Produtos Farmaceuticos SA (Sintra, Portugal).
Blood pressure, heart rate and drugs blood concentrations
Systolic blood pressure (SBP), diastolic blood pressure (DBP) and heart rate (HR) were measured before the animal sacrifice, by the non-invasive tail-cuff method, using a sphygmomanometer (LE 5001 Pressure meter, Letica Scientific Instruments, Spain). CsA blood concentrations were assessed by immunoassays using automatic methods (Flex reagent) and equipment (DimensionRRxL, Siemens, Germany).
Sample collection and preparation
Before the treatments (T0) and at the end of the acute and chronic protocols, the rats were injected with intraperitoneal anaesthesia with 2 mg/Kg BW of a 2:1 (v:v) 50 mg/mL Ketamine (KetalarR, Parke-Davis, Pfizer Laboratories Lda, Seixal, Portugal) solution in 2.5% chlorpromazine (LargatilR, Rhone-Poulenc Rorer, Vitoria laboratories, Amadora, Portugal). Blood samples were immediately collected by venipuncture from the jugular vein in needles with no anticoagulant (for serum samples collection) or with appropriated anticoagulant (ethylenediamine tetraacid – EDTA) for blood cell analysis. At the final time, the rats were sacrificed by cervical dislocation, and the kidneys were immediately removed, weighted and stored in RNA-stabilizer reagent for gene expression determinations, frozen in nitrogen for lipid peroxidation assays and pre-fixed with formaldehyde for histopathological analysis.
T-lymphocyte content and activation
Fresh peripheral blood samples were collected into EDTA vacutainer tubes. The mononuclear cells were then isolated from the other blood cells by density gradient centrifugation (Histopaque-1077 and -1119, from Sigma-Aldrich, Sintra, Portugal). The next steps, samples stainings and flow cytometry analysis, were described previously24.
Serum and urine data
Serum creatinine, blood urea nitrogen (BUN), glucose, total-cholesterol and high density lipoprotein (HDL), were evaluated by automatic validated methods and equipments (Hitachi 717 analyser, Roche Diagnostics Inc., MA, USA). Insulin levels were measured by using a rat ELISA kit from Mercodia (Uppsala, Sweden).
The animals were housed in metabolic cages during 24 hours and received tap water and food ad libitum. The urine concentration of creatinine and BUN were assessed in the 24 hour urine (Cobas Integra 400 plus, RocheR), and the urine volumes were measured in order to calculate creatinine and BUN clearance and the glomerular filtration rate, according to previously described protocols25.
Serum, kidney and 24 hours urine lipid peroxidation
Lipid peroxidation was determined by assaying the malondialdehyde (MDA) production by means of the thiobarbituric acid TBA) test, as previously described26.
RT-qPCR kidney gene expression
The kidney, was stored in RNA later solution (Ambion, Austin, TX, USA). For RNA extraction, 10 mg of tissue were weighted and 450 μL of RLT Lysis Buffer was added, tissue disruption and homogenization for 2 min at 30 Hz was performed using a TissueLyser (Qiagen, Hilden, Germany). Tissue lysates were processed according to the RNeasyR Mini Kit protocol (Qiagen, Hilden, Germany). Total RNA was eluted in 50 μL of RNase-free water (without optional treatment with DNAse). In order to quantify the amount of total RNA extracted and to verify RNA integrity (RIN, RNA Integrity Number), samples were analyzed using a 6000 Nano ChipR kit, in the Agilent 2100 bioanalyzer (Agilent Technologies, Walbronn, Germany) and the 2100 expert software, following manufacturers instructions. The isolation yield was from 0.5 to 3 μg; RIN values were 6.0–9.0 and purity (A260/A280) was 1.8–2.0. RNA was reverse transcribed with SuperScript III First-Strand Synthesis System for RTPCR (Invitrogen, California, USA). One microgram of total RNA was mixed with a 2× First-Strand Reaction Mix and a SuperScript III Enzyme Mix (Oligo(dT) plus Random hexamers). Reactions were carried out in a thermocycler Gene Amp PCR System 9600 (Perkin Elmer, Norwalk, CT, USA), 10 min at 25°C, 30 min at 50°C and 5 min at 85°C. Reaction products were then digested with 1 μL (2 U) RNase H for 20 min at 37°C and, finally, cDNA was eluted to a final volume of 50 μL and stored at −20°C. Gene expression was performed using a 7900 HT Sequence Detection System (Applied Biosystems, Foster City, USA). A normalization step preceded the gene expression quantification, using geNorm Housekeeping Gene Selection kit for Rattus norvegicus (Primer Design, Southampton, UK) and geNorm software (Ghent University Hospital, Center for Medical Genetics, Ghent, Belgium) to select optimal housekeeping genes for this study27. Real-time PCR reactions used specific QuantiTect Primer Assays (Qiagen, Hilden, Germany) with optimized primers for TGF- β1 (QT00187796), proliferating cell nuclear antigen (QT00178647), mechanistic target of rapamycin (QT00180586) and monoclonal antibody Ki-67 (QT00450786) as proliferative markers. Endogenous controls were also used: glyceraldehyde-3-phosphate dehydrogenase (QT00199633), actin beta (QT00193473), topoisomerase I (QT01820861) together with a Quanti-Tect SYBR Green PCR Kit (Qiagen, Hilden, Germany) used according to manufacturers instructions. RT-qPCR reactions were carried out with: 100ng cDNA sample, primers (50-200 nM) and 1x QuantiTect SYBR Green PCR Master Mix. Non template control reactions were performed for each gene, in order to assure non unspecific amplification. Reactions were performed with the following thermal profile: 10 min. at 95°C plus 40 cycles of 15 seconds at 95°C and 1 min. at 60°C. Real-time PCR results were analyzed with SDS 2.1 software (Applied Biosystems, Foster City, USA) and quantification used the 2- ΔΔCt method28. The results were obtained in CNQR (calibrated normalized relative quantities) and then converted in percentage with the control group as reference.
Histopatologica analysis
Staining techniques: Samples were fixed in Bocks fixative and embedded in paraffin wax, and 4 μm thick sections were stained with routine histopathological diagnosis techniques: Haematoxylin and Eosin (H&E); Periodic Acid of Schiff (PAS) and Massons Trichrome. All samples were examined by light microscopy using a Zeiss Microscope Mod. Axioplan 2. The degree of injury visible by light microscopy was scored in a double-blinded fashion by two independent pathologists. Lesions were evaluated on the total tissue on the slide.
Analysis of lesions.Glomerular damage was assessed by evaluating mesangial expansion, the glomerular basement membrane, the capsule of Bowman thickening, nodular sclerosis, glomerulosclerosis, atrophy, and hyalinosis of the vascular pole. The analysed tubulointerstitial lesions comprised inflammation, presence of hyaline cylinders, tubular basement membrane irregularity, tubular calcification, and the association of interstitial fibrosis and tubular atrophy (IFTA). The evaluation of vascular lesions was concentrated on arteriolar hyalinosis, arteriolosclerosis and arteriosclerosis. A semi-quantitative rating for each slide ranging from normal (or minimal) to severe (extensive damage) was assigned to each component.
Severity was graded as absent/normal (0), mild (1), moderate (2), and severe (3). Scoring was defined according to the extension of the lesion (number of capsules): normal: 0%; mild: <25%; moderate: 25–50%; severe: >50%. The final score of each sample was obtained by the average score observed in the individual glomeruli, in the considered microscopic fields.
Tubular calcification was evaluated and graded by the same semiquantitative method. Regarding vascular lesions, arteriosclerosis was scored as 0 if no intimal thickening was present, as 1 if intimal thickening was less than the thickness of the media, and as 2 if intimal thickening was more than the thickness of the media and considering the worst artery on the slide.
Using PAS, the rating was set for intensity and extension of staining, ranging from 0 (no staining) to 3 (intense and extensive staining), respecting tissue specificity scoring when adequate.
Statistical analysis
Statistical analyses were performed using the Graph-Pad PrismR for Windows (version 5.00). The results are presented as means ± S.E.M. Comparisons between groups were performed using the Students t-test.
Significance was accepted at p less than 0.05.
RESULTS
CsA blood concentration and T-lymphocyte activation
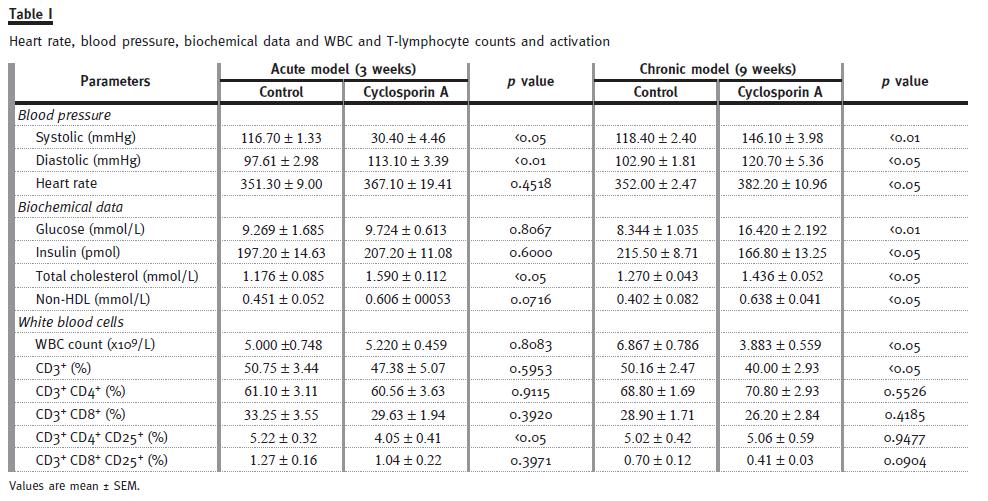
CsA dose that might mimic de trough concentration of CsA found in blood from humans under CsA immunosuppressive therapy was used in our rat model. Therefore, the trough blood concentration of CsA obtained using a 5 mg/kg/day dose in the rat (367.0 ± 45.5 ng/ml) was within the range achieved in humans. T-lymphocyte count was unchanged after 3 weeks of CsA treatment, however a significantly reduced (p<0.05) T-lymphocyte count was observed after 9 weeks (Table I), this was accompanied by a similar profile measured for the white blood cell (WBC) count. Moreover, activated CD4 T-lymphocytes showed a significantly reduced (p<0.05) number after 3 weeks of CsA exposure, while activated CD8 T-lymphocytes decreased after 9 weeks CsA treatment (Table I).
Biochemical data
Acute CsA treatment produced a significant (p<0.05) increase in the total cholesterol contents, without significant changes on glucose and insulin. However, longterm CsA-treatment aggravated total cholesterol and non-HDL levels (Table I). Furthermore, CsA promoted a significant rise in glucose (p<0.01) levels, contrasting with a significant (p<0.05) decrease in serum insulin.
Blood pressure and heart rate
CsA treatment induced a significant increase in both the systolic (p<0.05) and diastolic ( p<0.01) blood pressure (SBP and DBP) after just 3 weeks treatments.
The effect was even more pronounced in chronic therapy. Similar profile was encountered for the heart rate (HR), with a trend for increased values after the acute CsA exposure and a significantly increased value after the chronic treatment (p<0.05), suggesting an aggravation with a longer CsA exposure (Table I).
CsA-induced nephrotoxicity
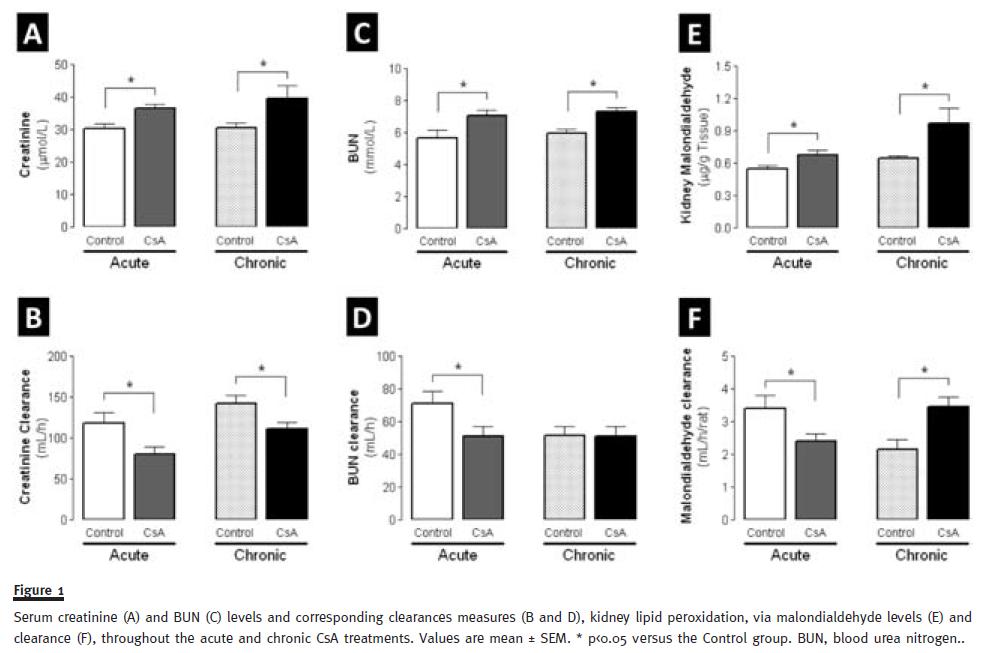
Serum creatinine and BUN levels significantly increased (p<0.05) in the acute CsA-treated group, accompanied by a significantly decreased (p<0.05) in creatinine and BUN clearances (Figure 1). Long-term CsA treatment further aggravated the serum creatinine and BUN levels, accompanied by a decrease in creatinine clearance(p<0.05). The glomerular filtration rate decreased both after 3 and 9 weeks of treatment (p<0.05) (data not shown). Concerning oxidative stress, acute treatment with CsA promoted significant increase in MDA levels (p<0.05), accompanied by MDA clearance decreased (p<0.05). Interestingly, chronic treatments of CsA, presented even more oxidative stress in kidney, with significant increased MDA clearance, contrasting with the decrease in acute model (Figure 1).
Despite the significantly increased markers of renal function (creatinine, BUN and MDA), acute CsA treatment was unable to promote significant morphological changes on the kidney tissue when compared with the control.
However, chronic CsA exposure induced important changes on the kidney (vessels, glomeruli and tubules) structure, suggesting important nephrotoxicity. The main changes encountered compared with the normal controls are represented in Table II. In the long-term CsA exposure, hyperaemia and vascular congestion were identified (significant versus Control) in the rat kidneys (data not shown), but arteriolosclerosis (p<0.05) and arteriolar vacuolization (p<0.01) were the main significant vascular lesions observed when compared with the control animals.
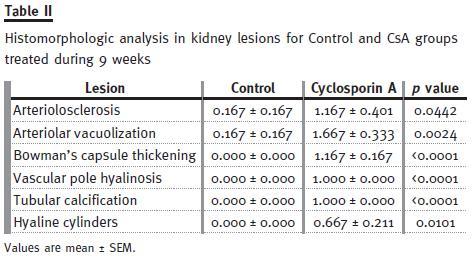
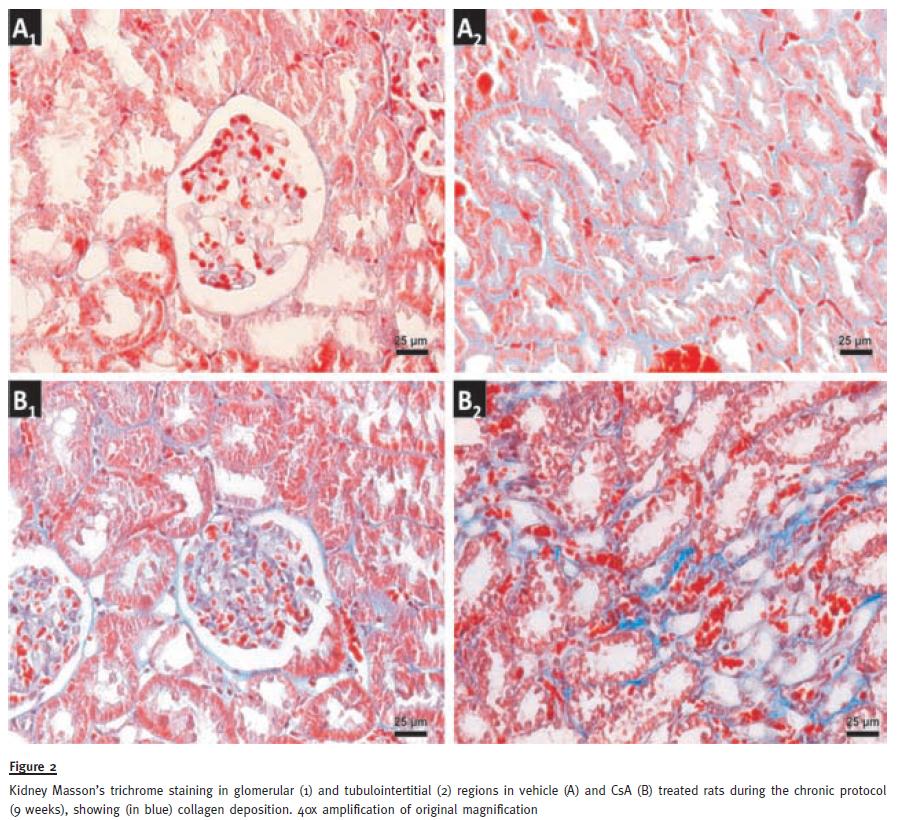
Regarding glomerular lesions, the major findings (p<0.05) were changes in mesangial expansion, hyalinosis of vascular pole and the thickening of Bowmans capsule (Table II), when compared with the control rat kidneys. In addition to tubular vacuolization, other tubular lesions were encountered, including hyaline cylinders, inflammatory infiltrate and tubular calcification, versus the normal profile found in the control rats. Figure 2 shows collagen deposition in glomerular and tubulointerstitial regions of chronic (9 weeks) CsA-treated rats (B1 and B2, respectively), when compared with normal Massons Trichrome staining in the control animals (A1 and A2).
Kidney gene expression data
Acute CsA treatment, produced a significant down-regulation of the antigen identified by the monoclonal antibody Ki67 (MKi67) (p<0.001), contrasting by a significant overexpression of proliferating cell nuclear antigen (PCNA) (p<0.001), transforming growth factor beta 1 (TGF-β1) (p<0.05) and mammalian target of rapamycin (mTOR) mRNA levels. On the other hand, in the chronic CsA treatment, PCNA and TGF-β1 expression were unchanged versus the control, while Mki67 (p<0.001) and mTOR (p<0.01) were remarkably overexpressed at this time, contrasting with the levels that these genes presented in the acute treatment (Figure 3).
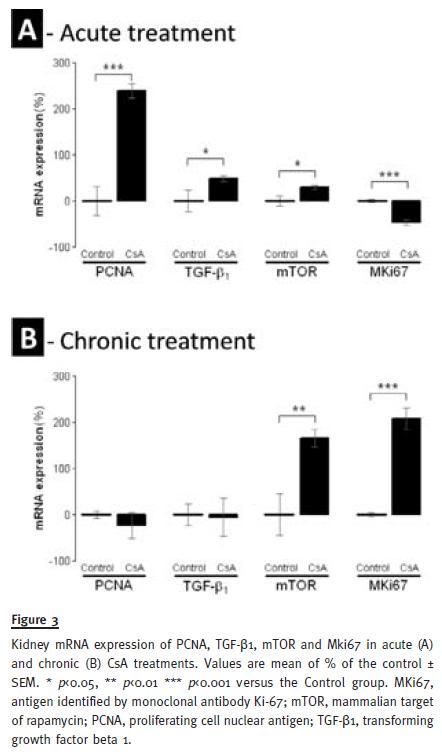
DISCUSSION
In the last decades, there has been a clear change in attitude to transplantation, with particular attention to the long-term kidney function and overall quality of life improvement. Despite the alternative therapeutics that has been sought, calcineurin inhibitors remain our most effective and widely used immunosuppressive agents29. In this context, the search for the precise molecular mechanism underlying acute vs chronic CsA-induced nephropathy is essential, and it depends largely on experimental studies. Our study was performed in order to compare, in a rat model, the acute versus chronic CsAnephrotoxicity (3 and 9 weeks, respectively), based on serum and urine markers, kidney histopathological evaluation and gene expression markers.
Concerning the immunosuppressive efficacy of the CsA treatments, we found that the short period administration was associated with unchanged values of WBC and T-lymphocyte, but the value of activated T-helper cells (CD4+CD25+), was significantly reduced. On the other hand, chronic CsA exposure was associated with a significant reduction of both WBC and T-lymphocyte counts (CD3+ cells), with a trend for a decrease in activated cytotoxic T cell (CD8+CD25+) count. It is known that CsA is very effective in inhibiting the production of interleukin-2 (IL-2), a soluble lymphokine known to amplify cytotoxic T cell responses, and it is also capable of preventing IL-2 receptor expression on the precursor cytotoxic T lymphocyte30,31, which is in agreement with our data (not shown). Our data support the idea that longer CsA exposure is associated with better immunosuppressive efficacy, at least when analysed in terms of T-lymphocyte population available.
Our group has previously demonstrated, using the same CsA dose in the rats, that some of the serious side-effects of CsA, including hypertension and dyslipidaemia, are already present after just 2 weeks of treatment18,32. The present work reinforces these data, additionally demonstrating that the effects were aggravated in a long-term treatment, which includes changes in lipid profile and glucose/insulin metabolism, as expected from clinical data, an augmented toxicity with increased duration of exposure. A similar profile was observed for hypertension and tachycardia development, which are also aggravated over time. Although tachycardia has been attributed to an overactivation of the sympathetic nervous system18,33, the mechanisms underlying CsA-induced hypertension have been described as multifactorial.
Hypertension has been pointed as cause of systemic vascular and platelet hyper-reactivity, by a reduction in vasodilator markers, such as nitric oxide (NO) and prostacyclin, and by an increase in vasoconstrictor markers, such as ET-1, TXA2, 5-HT or noradrenaline17,18,32-34. Additionally, activation of the reninangiotensin-aldosterone system and oxidative stress have also been identified as contributors to the hypertensive effects observed34,35. However, several lines of evidence suggest that the renal vasomotor disequilibrium could be the main origin of both hypertension and nephropathy36,37.
Regarding renal biochemical characterization, we found that the serum markers of renal dysfunction (creatinine and BUN) were already significantly increased after 3 weeks of treatment, and were further aggravated with prolonged CsA exposure. Similar variation was found for kidney tissue lipid peroxidation, and most importantly, for the analysis of renal tissue lesions. Therefore, despite increased serum creatinine and BUN, accompanied by decreased clearances of creatinine, BUN, and MDA, as well as the glomerular filtration rate after the third week of CsA therapy, very slight changes on tissue structure were found, with absent or only low grade of lesion. However, after the chronic CsA exposure, significant glomerular, tubular and vascular lesions were observed. As in clinical practice, renal markers (GFR, creatinine and BUN contents and clearances), were confirmed altered in CsA treatment. Additionally, we observed an interesting variation in biomarker levels between acute and chronic CsA treatment. The significant increase in the MDA clearance observed in our model raises some questions. In 1989, Knight and colleagues detected high MDA levels in urine of transplanted patients38, but they were unable to find the cause/effect of these observations, suggesting that it could be from: (a) renal lipid peroxidation directly related to the cyclosporine/azathioprine therapy, (b) drug-induced or other nephrotoxicity by an alternative mechanism with secondary lipid peroxidation, (c) increased lipid peroxidation owing to an immunologic response to the kidney graft, or (d) a combination of these possibilities. Our data suggests that MDA clearance could be a predictive CsAnephrotoxicity marker, as increased MDA clearance appears at the same time-point as the first kidney lesions. Oxidative stress can promote the formation of a variety of vasoactive mediators39 that can affect renal function directly by causing renal vasoconstriction or decreasing the glomerular capillary ultrafiltration coefficient, thus reducing the glomerular filtration rate. The current diagnostic techniques employed to detect CsA nephrotoxicity are still inadequate. The estimated GFR can be an insensitive indicator, since it depends on various factors, including age and blood factors. Creatinine and BUN clearances have poor diagnostic value40. Moreover, the relationship between proteinuria and CsA-nephrotoxicity is complex, limiting its power as an early indicator of CsA-nephrotoxicity20. Lipid peroxidation occurs as a result of multi-unsaturated lipids reacting with oxidizing agents, promoting oxidative stress in the kidney structures. Urinary MDA reflects the presence of renal damage, which may be the cause or the consequence of lipid peroxidation. Therefore, the correlation between MDA clearance and kidney lesion grade could be a good strategy to identify early CsA nephrotoxicity.
CsA immunosuppressive mechanism of action is deeply related with inhibition of transcriptional factors, including the NFAT, a CsA-sensitive transcription factor implicated in cytokine gene induction, which will be essential for IL-2 formation and further maturation of T-lymphocytes, needed for the immune response41. However, the NFAT isoforms are not T-cell specific, and inhibition of this pathway by CsA gives rise to toxicity beyond immunosuppression42.
However, not much is yet known concerning the influence of acute and chronic CsA exposure on kidney gene expression, particularly of several important mediators of fibrosis and proliferation, which are key pathways in the deterioration of renal tissue.
In relation to the molecular mechanisms involved in the short-term versus long-term nephrotoxicity, several previous reports suggest that severe glomerular and tubular lesions, including atrophy, fibrosis, inflammation and sclerosis, are linked to important mediators, such as an increased inflammatory response, with activation of NF-KB and increased TGF-β1 release, which then causes nephropathy mediated by fibrosis and apoptosis of renal cells43-45.
However, old and new versions of the putative mechanisms have been suggested, but the main question remains unsolved21. In our animal model, the acute CsA treatment was mainly associated with up-regulation of mediators of fibrosis and proliferation, with overexpression of TGF-β1 and PCNA. However, these changes could be accompanied by putative compensatory response (NF-κβ and TP53 appeared up-regulated – not shown), because no lesions were present in the acute CsA treatment, perhaps because the nuclear factor MKi67 was downregulated.
During prolonged CsA exposure, nevertheless, nephrotoxicity evolves, as viewed by the degree of increased histological lesions, which seems to be associated with other molecular pathways and mediator alterations. In fact, there was a significant overexpression o MKi67, contrary to what was observed after the acute treatment, accompanied by normalization in the counter-regulatory responses (NF-κβ and TP53). Moreover, this normalization in the compensatory response promoted an increase in mTOR expression, a serine/threonine protein kinase, important in regulating cell growth, proliferation, motility, survival, protein synthesis and transcription43. Complete restoration of renal morphology and function can occur after acute kidney injury induced by ischemic or toxic injury. Renal regeneration depends, in part, on the ability of the remaining viable tubular cells to proliferate and restore the injured tubular epithelium47. As Lieberthal et al (2009) demonstrated, mTOR plays an important role in mediating the process of regeneration and recovery, depending on the kidney damage extension48. Moreover, mTOR activity is low or absent in the normal kidney but increases markedly after acute kidney injury. In agreement, mTOR inhibition has been associated with amelioration of kidney fibrosis, glomerulosclerosis and interstitial inflammation, having an important role in renal disease49,50. Concerning transplantation, mTOR inhibitors have been used to replace CsA, because of its reduced putative side-effects, including nephrotoxicity10,11. Our results reinforce the rationale for the early substitution of CsA by mTOR inhibitors, not only because longer CsA exposure is notoriously more deleterious, promoting structural kidney deterioration, but also because mTOR overexpression seems to be a feature of the chronic CsA exposure. Recently, Luo et al (2013), showed that rapamycin is less fibrogenic than CsA.
Moreover, in chronic kidney disease, rapamycin also slows the progression of renal fibrosis and delays the onset of renal failure, trough reduction of glomerular hypertrophy, decreases proinflammatory and profibrotic cytokines production and decline interstitial inflammation48. Future studies should focus on the correlation between kidney gene expression and protein levels, as well as on validation of better putative markers of acute (preventable) nephrotoxicity, including easily accessible biological samples, such as the blood and urine, which can be achieved by satisfactory correlation with molecular changes in the kidney tissue.
In conclusion, CsA-induced nephrotoxicity is significantly aggravated over time and distinct mechanisms seem to underlie either acute or chronic nephropathy, being mTOR a key for renal damage. These findings reinforce two key aspects in post-transplant therapeutics: (1) the continuous need to better identify early CsA nephrotoxicity markers and (2) the justification for early substitution of CsA by other less nephrotoxic immunosuppressive agents, with mTOR inhibitors a validated choice, in order to reduce the risk of chronic allograft nephropathy, thus improving outcomes in transplanted patients.
References
1. Halloran PF. Immunosuppressive drugs for kidney transplantation. N Engl J Med 2004;351:2715-2729 [ Links ]
2. Colombo D, Ammirati E, Cyclosporine in transplantation – a history of converging timelines. J Biol Regul Homeost Agents 2011;25:493-504 [ Links ]
3. Chapman JR. Chronic calcineurin inhibitor nephrotoxicity-lest we forget. Am J Transplant 2011;11:693-697 [ Links ]
4. Ponticelli C, Cucchiari D, Graziani G. Hypertension in kidney transplant recipients. Transpl Int 2011;24:523-533 [ Links ]
5. Bremer S, Vethe NT, Rootwelt H, et al . Mycophenolate pharmacokinetics and pharmacodynamics in belatacept treated renal allograft recipients – a pilot study. J Transl Med 2009;7:64 [ Links ]
6. Textor SC, Taler SJ, Canzanello VJ, et al. Posttransplantation hypertension related to calcineurin inhibitors. Liver Transpl 2000;6:521-530 [ Links ]
7.Cattaneo D, Perico N, Gaspari F et al. Nephrotoxic aspects of cyclosporine. Transplant Proc 2004;36:234S-239S [ Links ]
8. Mange KC, Cizman B, Joffe M, et al. Arterial hypertension and renal allograft survival. JAMA 2000;283:633–638 [ Links ]
9. Barbari AG, Stephan AG, Masri MA. Calcineurin inhibitor-free protocols: risks and benefits. Saudi J Kidney Dis Transpl 2007;18:1-23 [ Links ]
10. Mota A. Sirolimus: a new option in transplantation. Expert Opin Pharmacother 2005;6:479-487 [ Links ]
11. Campistol JM, Cockwell P, Diekmann F, et al. Practical recommendations for the early use of m-TOR inhibitors (sirolimus) in renal transplantation. Transpl Int 2009;22:681-687 [ Links ]
12. Marti HP, Frey FJ. Nephrotoxicity of rapamycin: an emerging problem in clinical medicine. Nephrol Dial Transplant 2005;20:13-15 [ Links ]
13. Boraty ńska M, Banasik M, Watorek E, et al. Conversion to sirolimus from cyclosporine may induce nephrotic proteinuria and progressive deterioration of renal function in chronic allograft nephropathy patients. Transplant Proc 2006;38:101-104 [ Links ]
14. Rangan GK. Sirolimus-associated proteinuria and renal dysfunction. Drug Saf 2006;29:1153-1161 [ Links ]
15. Bunnapradist S, Vincenti F. Transplantation: To convert or not to convert: lessons from the CONVERT trial. Nat Rev Nephrol 2009;5:371-373 [ Links ]
16. Mota A, Arias M, Taskinen EI, et al. Rapamune Maintenance Regimen Trial. Sirolimusbased therapy following early cyclosporine withdrawal provides significantly improved renal histology and function at 3 years. Am J Transplant 2004;4:953-961 [ Links ]
17. Reis F, Tavares P, Rito LC, et al. Platelet activation is increased in cyclosporin A-induced hypertensive rats. J Cardiovasc Pharmacol 2000;36:56-64 [ Links ]
18. Reis F, Rocha L, Ponte L, et al. Effect of preventive and regressive isosorbide 5-mononitrate treatment on catecholamine levels in plasma, platelets, adrenals, left ventricle and aorta in cyclosporin A-induced hypertensive rats. Life Sciences 2005;77:2514-2528 [ Links ]
19. Reis F. The unsolved cyclosporine-induced kidney injury: is paricalcitol a feasible new renoprotective option?. Kidney Int 2010;77:1055-1057 [ Links ]
20. OConnell S, Slattery C, Ryan MP, et al. Identification of novel indicators of cyclosporine A nephrotoxicity in a CD-1 mouse model. Toxicol Appl Pharmacol 2011;252:201-210. [ Links ]
21. Sarro E, Jacobs-Cacha C, Itarte E, et al. A pharmacologically-based array to identify targets of cyclosporine A-induced toxicity in cultured renal proximal tubule cells. Toxicol Appl Pharmacol 2012;258:275-287 [ Links ]
22. Lesko LK, Atkinson Jr AJ. Use of biomarkers and surrogate endpoints in drug development and regulatory decision making: criteria, validation, strategies. Annu Rev Pharmacol Toxicol 2001;41:347-366 [ Links ]
23. Jiang Y, Gerhold DL, Holder DL, et al. Diagnosis of drug-induced renal tubular toxicity using global gene expression profiles. Journal of Translational Medicine 2007;5:47 [ Links ]
24. Sereno J, Parada B, Rodrigues-Santos P, et al. Serum and renal tissue markers of nephropathy in rats under immunosuppressive therapy: cyclosporine versus sirolimus. Transplant Proc 2013;45:1149-1156 [ Links ]
25. Pestel S, Krzykalla V, Weckesser G. Measurement of glomerular filtration rate in the conscious rat. Journal of Pharmacological and Toxicological Methods 2007;56:277–289 [ Links ]
26. Piloto N, Teixeira HM, Teixeira-Lemos E et al, Erythropoietin promotes deleterious cardiovascular effects and live risk in a rat model of chronic sports doping. Cardiovasc Toxicol 2009;9:201-210 [ Links ]
27. Vandesompele J, De Preter K, Pattyn F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 2002;3:RESEARCH0034 [ Links ]
28. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2- ΔΔCT method. Methods 2001;25:402–408 [ Links ]
29. Gaston RS. Chronic calcineurin inhibitor nephrotoxicity: reflections on an evolving paradigm. Clin J Am Soc Nephrol 2009;4:2029-2034 [ Links ]
30. Colombani PM, Hess AD. T-lymphocyte inhibition by cyclosporine. Potential mechanisms of action. Biochem Pharmacol 1987;36:3789-3793 [ Links ]
31. Mattila PS. The actions of cyclosporin A and FK506 on T-lymphocyte activation. Biochem Soc Trans 1996;24:45-49 [ Links ]
32. Reis F, Tavares P, Fontes Ribeiro CA, et al. The peripheral serotonergic system and platelet aggregation in cyclosporin A-induced hypertensive rats. Thromb Res 1999;96:365-372 [ Links ]
33. Sander M, Lyson T, Thomas GD, et al. Sympathetic neural mechanisms of cyclosporineinduced hypertension. Am J Hypertens 1996;9:121S-138S [ Links ]
34. Reis F, Rocha-Pereira P, Teixeira de Lemos E, et al. Oxidative stress in cyclosporineinduced hypertension: evidence of beneficial effects or tolerance development with nitrate therapy. Transplant Proc 2007;39:2494-2500 [ Links ]
35. Lassila M. Interaction of cyclosporine A and the renin-angiotensin system; new perspectives. Curr Drug Metab 2002;3:61-71 [ Links ]
36. Vaziri ND, Ni Z, Zhang YP, et al. Depressed renal and vascular nitric oxide synthase expression in cyclosporine-induced hypertension. Kidney Int 1998;54:482-491 [ Links ]
37. Zhang W, Victor RG. Calcineurin inhibitors cause renal afferent activation in rats: a novel mechanism of cyclosporine-induced hypertension. Am J Hypertens 2000;13:999-1004 [ Links ]
38. Knight JA, Cheung AK, Pieper RK, et al. Increased urinary lipoperoxide levels in renal transplant patients, Ann Clin Lab Sci 1989;19:238-241 [ Links ]
39. Garcia-Cohen EC, Marin J, Diez-Picazo JD, et al. Oxidative stress induced by tert-butyl hydroperoxide causes vasoconstriction in the aorta from hypertensive and aged rats: role of cyclooxygenase-2 isoform. J Pharmacol Exp Ther 2000;93:75-81 [ Links ]
40. Dieterle F, Perentes E, Cordier A, et al. Urinary clusterin, cystatin C, beta2-microglobulin and total protein as markers to detect drug-induced kidney injury. Nat Biotechnol 2010;28:463-469 [ Links ]
41. Rao A. NFATp, a cyclosporin-sensitive transcription factor implicated in cytokine gene induction. J Leukoc Biol 1995;57:4:536-542 [ Links ]
42. Liu EH, Siegel RM, Harlan DM, et al. T cell-directed therapies: Lessons learned and future prospects. Nat Immunol 2007;8:25–30 [ Links ]
43. Naesens M, Kuypers DR, Sarwal M. Calcineurin inhibitor nephrotoxicity. Clin J Am Soc Nephrol 2009;4:481-508 [ Links ]
44. Li C, Yang CW. The pathogenesis and treatment of chronic allograft nephropathy. Nat Rev Nephrol 2009;5:513-519 [ Links ]
45. Park JW, Bae EH, Kimet IJ, et al. Paricalcitol attenuates cyclosporine-induced kidney injury in rats. Kidney Int 2010;77:1076-1085 [ Links ]
46. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012;149:274-293 [ Links ]
47. Nigam S, Lieberthal W. Acute renal failure: III. The role of growth factors in the process of renal regeneration and repair. Am J Physiol Renal Physiol 2009;279:F3–F11 [ Links ]
48. Lieberthal W, Levine JS. The role of the mammalian target of rapamycin (mTOR) in renal disease. J Am Soc Nephrol 2009;20:2493-2502 [ Links ]
49. Kramer S, Wang-Rosenke Y, Scholl V, et al. Low-dose mTOR inhibition by rapamycin attenuates progression in anti-thy1-induced chronic glomerulosclerosis of the rat. Am J Physiol Renal Physiol 2008;294:F440-F449 [ Links ]
50. Chen G, Chen H, Wang C, et at. Rapamycin ameliorates kidney fibrosis by inhibiting the activation of mTOR signaling in interstitial macrophages and myofibroblasts. PLoS One 2012;7:e33626 [ Links ]
Professor Dr. Flavio Reis*, PhD
Laboratory of Pharmacology and Experimental Therapeutics, IBILI,
Medicine Faculty, Sub -Unit 1 (Polo III), Coimbra University,
3000 -548 Coimbra, Portugal
E-mail: freis@fmed.uc.pt
Acknowledgments: This work was supported by the PhD research grant from the Portuguese Foundation for Science and Technology (SFRH/BD/63962/2009) and Strategic Project PEst-C/SAU/UI3282/2011-COMPETE, PTDC/SAU-OSM/104124/2008. This study had the kind collaboration of Novartis Pharma (Lisbon, Portugal) who supplied the CsA (Sandimmune Neoral).
Conflict of interest statement: The authors declare no conflict of interest.
Received for publication: 28/06/2012
Accepted in revised form: 30/08/2013


{kind=link}
{kind=link}
{kind=link}