










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
versão impressa ISSN 0872-0169
Port J Nephrol Hypert vol.28 no.3 Lisboa set. 2014
EDITORIAL
Soluble endoglin: a biomarker or a protagonist in the pathogenesis of preeclampsia?
Lucia Perez -Roque, Jose M. Lopez –Novoa
Unidad de Fisiopatología Renal y Cardiovascular; Departamento de Fisiología y Farmacología, Universidad de Salamanca. 37007 Salamanca, Spain.
Instituto de Investigaciones Biomédicas de Salamanca, (IBSAL), 37007 Salamanca, Spain
Instituto Reina Sofía de Investigación Nefrológica, Fundación Renal Iñigo Alvarez de Toledo, 28034 Madrid, Spain
INTRODUCTION
Pre-eclampsia is a pregnancy-specific disorder characterized by endothelial dysfunction, hypertension (> 140/90 mmHg) and proteinuria (> 300mg/24hours) in the third trimester of pregnancy. It affects about 3 -5% of all pregnancies worldwide1,2. It remains a major cause of maternal mortality (15–46%) and is responsible for a 5 -fold increase in perinatal mortality3.
The World Health Organization has recognized the importance of pre-eclampsia by launching a global programme to fight pre-eclampsia-eclampsia (World Health Organization. Policy and Coordination Committee 16th meeting. Department of Reproductive Health and Research. Summary Medium-term Programme of Work 2004 -2009 HRP/ PCC (16)/2003/8.1).
In addition, women who have suffered pre eclampsia and their offspring are at greater risk of developing cardiovascular disease later in life4.
Several risk factors are associated for pre-eclampsia; these include nulliparity, multifetal gestations, previous history of pre-eclampsia, obesity, diabetes mellitus, vascular and connective tissue disorders like systemic lupus erythematosus and antiphospholipid antibodies. Severe pre-eclampsia can lead to the appearance of the HELLP (haemolysis, elevate liver function tests, low platelets) syndrome, the most critical variant of pre -eclampsia, and is associated with a higher risk of maternal and neonatal adverse outcomes5.
At the present, there are no effective treatments for this disease, and delivery remains the only solution for maternal symptoms. In many cases, the newborn is often premature and there are significant perinatal morbidity and mortality among such babies.
Until recently, the molecular pathogenesis of pre-eclampsia was largely unknown, but recent observations support the hypothesis that altered expression of placental anti -angiogenesis factors are responsible for the clinical manifestation of this disease. Two anti -angiogenic factors soluble fms -like kinase 1 (sFlt1) and soluble endoglin (sEng) have been reported to have increased plasma concentration in the maternal circulation weeks before the onset of pre-eclampsia6,7 and decreased quickly after delivery8. The concentration of these factors in plasma is correlated with the severity of pre -eclampsia and bad prognossis. Thus, they have been proposed as useful biomarkers for the diagnostic and prognosis of pre-eclampsia9,10.
Also, Gilbert et al showed that sEng and sFlt1 were increased in the serum of uteroplacental ischaemia animal model (RUPP)11,12. sFtl1 is a soluble form of the VEGF receptor-1, a cell membrane receptor for VEGF (vascular endothelial growth factors) and PIGF (placental growth factor -1), whereas sEng is the soluble form for a membrane co -receptor of several members of the TGF β superfamily. Both, sFlt1 and sEng, are secreted by the placenta13,14.
PLACENTA IS THE ORIGIN OF PRE -ECLAMPSIA
Placenta plays a central role in pre-eclampsia as it is evidenced by rapid disappearance of disease symptoms after delivery and maintenance of these symptoms when placenta is retained. Abnormalities in the placental development have been studied in order to understand the early stage of this disease. The molecular basis for placental dysregulation of pathogenic factors it is not clear.
The first event in the early gestation of pre -eclampsia is the abnormal placentation by alterations in the placenta vascular remodeling15. It has been reported unappropriated vascular remodeling of uterine spiral arteries by impaired invasion of these arteries by trophoblasts. On this way, the blood supply to the placenta is reduced to a level unable of providing adequate placental perfusion to sustain the growing fetus16 and generates a hypoxic environment. In humans, trophoblasts stem cells are differentiated into two cell types, villous trophoblasts and extravillous trophoblasts (EVTs). Earlier in pregnancy, invading trophoblasts, called interstitial EVTs, migrate into the decidualized endometrium and endovascular EVTs migrate along the lumen of spiral arterioles17. This EVT invasion creates the large -diameter and low-resistant vessels that carry blood to the placenta. After 12 weeks, endovascular EVTs invade the uterine spiral arteries, replace the endothelial cells, and participate in the degradation of tunica media smooth muscle cells. This remodeling of the spiral arteries is essential for proper placental perfusion to sustain fetal growth. Pre -eclampsia is considered to be caused by poor placentation that can be explained by the shallow invasion by trophoblast until 20 weeks of gestation. This impairment is closely related with a failure of vascular remodeling. The consequence is shallow spiral arteries that contribute to the hypoxic environment in the placenta18,19. Therefore, the hypoxic environment has been postulated as a critical signal that initiates the pathogenic process of pre-eclampsia20. There are a lot of evidence supporting the central role of hypoxia in pre -eclampsia. It was reported that the placentas of pre-eclamptic women and models of placental hypoxia show similar gene expression21. In addition, reductions in utero placental blood flow in animal models induced hypertension, proteinuria and endothelial dysfunction, which are the typical sign of this disease22. This relationship between uteroplacental ischaemia and pre-eclampsia was demonstrated in several animals, such as dogs23, rabbits24,25, primates26,27 and, more recently, it has been established a reduced uterine perfusion pressure model in the pregnant rat (RUPP) in which the utero placental perfusion is reduced by approximately 40% and shows many features of pre-eclampsia28-32.
The possibility that sFlt1 and sEng are a key pathogenic link between placental pathology and maternal endothelial damage provides hope that these substances, in addition to serving as biomarkers of pre-eclampsia, could also play some role in the pathogenesis of the disease and, thus, could possibly also be effective therapeutic targets for the disease. In this editorial we will focus on sEng, in order to analyze if it is only a biomarker or if it could also be a protagonist in the pathogenesis of pre-eclampsia.
ENDOGLIN
Endoglin (Eng), also called CD105, is a glycosylated transmembrane protein that acts as an auxiliary receptor of several members of the TGF -β superfamily of proteins13.
Endoglin is highly expressed in endothelial cells and syncytiotrophoblast33,34. Although the most studied physiological effect of Eng is regulating angiogenesis [1], we have reported that Eng plays a major role in the regulation of vascular tone13. It has been described that the vasodilator effect of Eng is mediated by regulating endothelial nitric oxide synthase (eNOS) expression and NO synthesis35. It has been proposed that Eng plays a major role in pre-eclampsia36. In this review, we will describe the mechanisms of Eng overexpression and sEng release in pre-eclampsia, as well as the possible role of sEng in hypertension, proteinuria and abnormal placentation, all of them characteristic of pre-eclampsia.
Mechanisms of placental endoglin release in pre-eclampsia Hypoxia seems to be involved in increased endoglin expression in the placenta
Endoglin is overexpressed in placenta during pre-eclampsia. Eng mRNA expression in chorionic villi at 11-week's gestation were significantly higher in women that will develop pre -eclampsia. These findings suggest that this increase could be implicated in the early pre -eclampsia37. The development of the placenta during the early normal pregnancy is carried out in an environment of low oxygen tension until 10 or 12 weeks of gestation. In that moment, the intravillous space is filled to maternal blood and the oxygen levels begin to increase. High level of endoglin is found in the early stage of gestation when the oxygen is low, but in the later stage the levels of endoglin is decreased38,39. However, in pre -eclamptic placentas the amount of endoglin is always elevated. In trophoblasts obtained from placentas of pre-eclampsia patients, the release of sEng in hypoxia conditions (2% O2 tension) was also higher than in trophoblast from normal pregnant women40. These facts confirm that the hypoxic environment induces endoglin expression and plays an important role in the pathogenesis of pre-eclampsia41 -43. The mechanism explaining high endoglin expression in the presence of placentary hypoxia is based in the fact that endoglin gene promoter has a hypoxia responsive element (HRE) region. HIF -1, in cooperation with Sp1 and the coactivator CBP/P300 is bound to this region stimulating endoglin expression. This stimulatory effect is further enhanced when the HIF -α/Sp1 complex collaborates with the TGF β/Smad3 signaling pathway41.
On the other hand, there are data suggesting that the increase in Eng expression observed in pre-eclampsia can also be mediated, at least in part, by HIF -1 α, probably through activation of the TGF -β3 pathway43,44. Tal R. et al have demonstrated the effects of HIF -1 overexpression in pregnant mice using adenovirus which express a constitutively active and stabilized HIF -1α. They observed an elevation of sFlt1 and sEng in the plasma of these mice45.
Increased oxidative stress can be involved in increased Eng expression by the placenta
A crosstalk between hypoxia and oxidative stress has been described by several authors. Under hypoxic conditions, reactive oxygen species (ROS) are produced by a flavoprotein-containing NAD(P)H or by mitochondria as a mechanism that contributes to stabilize HIF -1α and consequently mediates the transcriptional response to hypoxia46. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor -1alpha during hypoxia, nuclear translocation and transcriptional activation by a mechanism of O2 sensing46. Thus, hypoxic conditions may favor the generation of ROS in pre-eclampsia. Indeed, oxidative stress has been shown to play a pivotal role in the etiopathogenesis of pre-eclampsia20. Furthermore, it has been demonstrated that a decrease in endogenous antioxidant factors, such as the antioxidant hemeoxygenase-1 (HO-1), superoxide dismutase and glutathione peroxidase, may contribute to the increase release of sEng47-49.
Several studies have demonstrated high levels of LDL in the serum from women with pre-eclampsia and these LDL show a higher susceptibility to oxidation giving oxidized LDL (oxLDL) rich in oxysterols50.
Endogenously oxygenated cholesterol derivatives, such as 24(S)-hydroxycholesterol or 22(R)–hydroxycholesterol (oxysterols), are detected at relatively high levels in the placenta51. Oxysterols, a product of cholesterol oxidation, can play a role mediating sEng release in pre-eclampsia. Oxysterols have been identified as endogenous agonists of liver X receptors (LXRs). LXRs are ligand-activated transcription factors, which have two isoform: LXRα and LXRβ LXR α is present in tissues with high lipid metabolism, such as liver, intestine, adipose tissue and kidney, whereas LXR β is ubiquitously expressed52. It has been suggested a role of LXRs in the reproductive system and a potential application of LXR agonists in the treatment of reproductive pathologies53. It has been reported the association of the LXR β activation and pre-eclampsia. Mouzart et al. showed that the single nucleotide polimorphisms rs2695121 in LXR β gene was strongly associated with pre-eclampsia. In contrast, they did not find a significant association between the polimorphisms in LXR α gen and this disease54. On the other hand, Henry-Berger et al. have shown that endoglin is one of the target gene of LXR. Endoglin has six putative response elements (LXRE) in its proximal promoter region. The heterodimer LXR/RXR is able to bind them and stimulate its transcription. It has been reported that endoglin was up-regulated by administration of LXR agonists in human choriocarcinoma JAR cells55. Therefore, the LXR activation is able to up -regulate the inappropriate expression and function of endoglin and contribute with the complication of pregnancy56.
Mechanisms involved in the release of sEng in the placenta
In pre-eclamptic women, the high levels of sEng seems be derived from membrane isoform of the placental cells14.The release of sEng occurs by proteolytic cleavage of the membrane isoform. It is known that this proteolytic cleavage is mediated by membrane metalloproteinases, which are increased in pregnancy. Hawinkels et al, reported that metalloproteinase -14 (MMP14 or MT1 -MMP) is involved in the release of sEng57. Kaituu -Lino et al. treated the syncitiotrophoblast cell line (BeWo) with metalloproteinase inhibitor, GM6001, and observed a significant decreased in sEng release. The GM6001 inhibits other MMPs besides MMP14, so they administered MMP -13 or MMP -2/9 specific inhibitors, and did not observe differences in sEng release compared with its controls58. Furthermore Kaituu -Lino, et al., also demonstrated that there are other metalloproteinase present in placenta, such as MMP-16,-24,-25, but they were not associated with pre-eclampsia.
Furthermore, MMP -17 and -15 were also up–regulated in pre-eclampsia, but neither of them were involved in the increased release of sEng59,60.
Recently, a study from our group demonstrated that hypoxia induces sEng shedding in cultured human trophoblasts (JAR cells) and this release was increased by ROS. This study also demonstrated that oxysterols are involved in the sEng shedding through LXR pathway and that oxysterols and activators of LXR pathway increase cell surface expression and activity of MMP14 and inhibit the TIM -3 expression which is a MMP14 inhibitor55. Thus, we can suggest that oxysterols induce sEng release by increasing cell surface endoglin56, and by increasing MMP14 and inhibit its inhibitor TIMP -355. Experiments in vivo have also demonstrated that administration of LXR agonist or oxysterols induced the increase in plasma levels of sEng in mice supporting the hypothesis that the LXR activation increases sEng release55.
On the other hand, Lamarca et al, in the uteroplacental ischaemia animal model (RUPP), have reported high levels of antibodies to angiotensin receptor type I, AT1 -AA. They also demonstrated that the increase of AT1 -AA induces the rise of TNF -α, which stimulates the release of sEng and sFlt161.
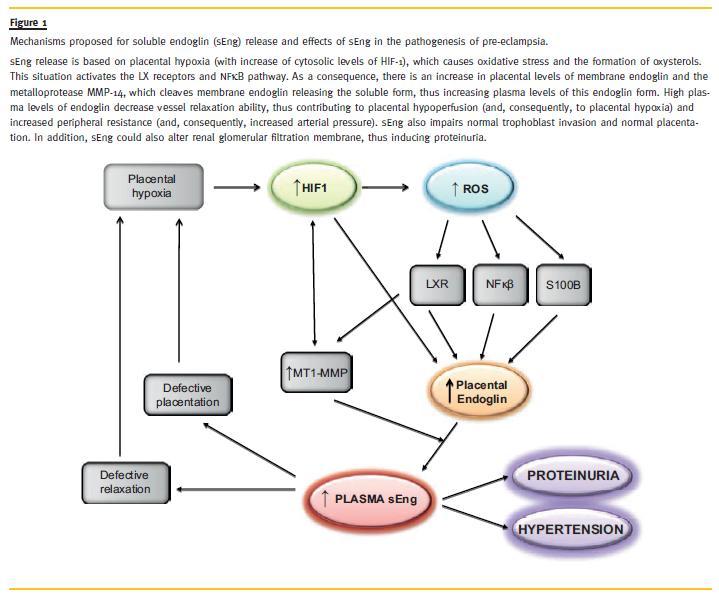
Thus, from our own results and those of the literature we can hypothesize that soluble endoglin release is based on placental hypoxia, which causes oxidative stress and the formation of oxysterols. As a consequence, there is an increase in placental levels of membrane endoglin and the metalloprotease MMp -14, which cleaves membrane endoglin releasing the soluble form, thus increase plasma levels of this form (Fig. 1).
Possible effects of endoglin in pre-eclampsia Effects of sEng in arterial pressure
The possible role of sEng in pre-eclampsia is not elucidated yet. It has been hypothesized that sEng plays its antiagiogenic and pro-hypertensive effects in pre -eclampsia due to its ability to bind circulating TGF β. TGF β, in the presence of membrane Eng, produce vasodilatation mediated by eNOS overexpression62.
Thus, the binding of TGF -β by sEng could prevent the interaction of these TGF β molecules with the cell -surface Eng and, consequently, its normal effects in the endothelium63. By this mechanism, sEng leads abolishes TGF β-dependent eNOS activation, thus leading to vasoconstriction14. Several studies reported that decreased NO availability is one of the principal causes of endothelial dysfunction in pre-eclampsia and is involved in haemodynamics changes characteristics of pre-eclampsia64,65. However, this hypothesis needs to be adequately confirmed.
Other authors have suggested that sEng alone cannot scavenge TGF β for which it has very low affinity, and suggest that sEng binds with high affinity to BMP9, which stimulates secretion from endothelial cells of the vascoconstrictor endothelin-1, also implicated in endothelial cell stabilization66. It remains to be determined if scavenging of circulating BMP9 by sEng is important in pre-eclampsia and regulation of hypertension. It should be noted that administration of LXR agonist or oxysterols, in addition to inducing the increase in plasma levels of sEng, also induces hypertension in mice, whereas when sEng release was prevented by a blocking peptide, hypertension was not observed55. Furthermore, male mice transgenic for sEng and with high plasma sEng levels also show hypertension55. In the RUPP model in rats, the hypertension is associated to high plasma levels of sEng11. Thus, all these data suggest that soluble endoglin plays a major role in the pathogenesis of pre -eclampsia–associated hypertension.
Effects of sEng in proteinuria
Classically, glomerular endothelial injury was considered responsible for the renal impairment present in pre-eclampsia. Proteinuria, another major characteristic of pre-eclampsia, has been explained by endotheliosis of glomerular capillaries, an alteration characterized by endothelial cell swelling and loss of endothelial fenestrae in these capillaries. It has been demonstrated that neutralization of TFG -β by neutralizing TGF-beta1 antibodies results in loss of glomerular fenestrae and proteinuria, thus suggesting that the glomerular endotheliosis lesions of pre-eclampsia are at least partially due to the placenta-derived sEng, which block the TGF -beta/endoglin signaling, respectively, causing the loss of glomerular endothelial cell fenestrae, cell swelling and proteinuria67.
Podocytes are modified glomerular epithelial cells that cover the outside surface of glomerular capillaries, and play a major role in regulating glomerular permeability to proteins. Recent evidence suggests that appearance of podocytes in urine is associated with the appearance and prognosis of pre-eclampsia68. There are some data suggesting that podocytes are crucial in explaining the loss of filtration capacity of the pre–eclamptic kidney69. Also, recent studies from our laboratory have demonstrated that sEng is able to prevent leukocyte binding to endothelium by interfering in the binding of leukocyte integrins to the RGD domain of endothelial endoglin70. Thus, it can be hypothesized that by a similar mechanism, sEng is able to reduce the binding of the podocytes to glomerular basal membrane (GBM) by impairing the binding of integrin α 3 β 1 in the podocyte to the GBM component laminin-521, one of the major mechanism for podocyte attachment71. This could induce the detachment of podocytes from the GBM, as these cells are subjected to a great force by the fluid that is filtered through the wall of the glomerular capillaries.
Effects of sEng in placentation
As above described, endoglin is greatly expressed in syncyotrophoblast and extravillous trophoblast (EVT). It was shown that the inappropriate levels of endoglin could induce a wrong trophoblast differentiation though a negative regulation by TGF β1 and TGFβ372. Studies have demonstrated that overexpression of endoglin decreased cell migration and invasion in mouse fibroblast or endothelial cells. Endoglin also has an important role in trophoblast biology43, through its ligands, TGF β1 and TGF β372. TGF β signaling is involved in several inhibitory signals, such as antiproliferative, antimigratory and anti -invasive signals.
Furthermore, it is demonstrated that LXR β activation reduced EVT invasion and migration through TGF β pathway73.
In addition, sEng can be also involved in inadequate placentation. It has been reported that sEng reduces the invasive ability of cytotrophoblasts through regulation of the expression of MMP-2 and MMP-9, metalloproteases necessary for a correct trophoblast invasion74. It has been reported that the invasion and vascular remodeling under hypoxia were significantly reduced in autophagy–deficient EVT cells compared with wild-type EVT cells. Interestingly, sEng suppressed EVT invasion by inhibiting autophagy. This effect of sEng was recovered by TGF -β1 treatment in a dose-dependent manner.
A high dose of sEng inhibited the vascular construction by EVT cells and endothelial cells, whereas a low dose of sEng inhibited the replacement of endothelial cells by EVT cells75. Although the evidences are very limited, we can hypothesize that sEng is involved in the inadequate placentation, which is the primary cause of pre-eclampsia. Thus, we can hypothesize, in the basis of the above described, that increased plasma levels of sEng are not only an early biomarker of pre-eclampsia but are also involved in the pathogenesis of the disease (defective placentation, hypertension and proteinuria) (Fig. 1)
CONCLUSION AND PERSPECTIVES
In summary, placenta plays the central role in pre-eclampsia. The first event in early gestation is an abnormal placentation by alterations in vascular remodeling. The triggering of the hypoxia pathway induces an oxidative stress in the placenta. The oxidative stress and high levels of cholesterol in pre-eclamptic women induce an increased production of oxysterols in maternal plasma. This leads to activation of the oxysterols receptors LXR, which induces sEng release, and increased plasma levels of sEng contributes to the clinical manifestation of pre-eclampsia: hypertension and proteinuria, and contributes to defective placentation, thus creating a vicious circle that amplifies the disease. A schematic representation of these mechanisms is shown in Fig. 1. According with these results, it can be speculated that one of the therapies for pre-eclampsia should be the reduction of sEng shedding. We have hypothesized that inhibition of the enzymatic activity of MMP14 could prevent sEng release. We have described a peptide (P583), which corresponds to the aminoacids 583-590 of endoglin sequence. This sequence includes the MMP14 cleavage site GL and interferes with its proteolytic activity. It was proven in vitro that P583 inhibits the sEng shedding. Administration of p583 to mice treated with LXR agonist or oxysterols reduced plasma levels of sEng and reverted the appearance of hypertension in these mice55. These studies suggest that a therapeutic strategy that inhibits sEng release or eliminates sEng from the circulation could be effective in preventing pre -eclampsia, a disease without treatment at present.
References
1. Bernab eu C, Lopez -Novoa JM, Quintanilla M. The emerging role of TGF -beta superfamily coreceptors in cancer. Biochim Biophys Acta 2009;1792(10):954-973. [ Links ]
2. Eiland E, Nzerue C, Faulkner M. Preeclampsia 2012. J Pregnancy 2012; doi: 10.1155/2012/586578. [ Links ]
3. Duley L. The global impact of pre -eclampsia and eclampsia. Semin Perinatol 2009;33(3):130-137. [ Links ]
4. Smith GC, Pell JP, Walsh D. Pregnancy complications and maternal risk of ischaemic heart disease: a retrospective cohort study of 129,290 births. Lancet 2001;357(9273):2002-2006. [ Links ]
5. Rachdi R, Fekih MA, Massoudi L, et al. [HELLP syndrome. Epidemiological, nosological and prognostic aspects]. Rev Fr Gynecol Obstet 1993;88(4):230-235. [ Links ]
6. Levine RJ, Lam C, Qian C, Yu KF, et al. Soluble endoglin and other circulating antiangiogenic factors in preeclampsia. N Engl J Med 2006;355(10):992 -1005. [ Links ]
7. Troisi R, Braekke K, Harsem NK, Hyer M, Hoover RN, Staff AC. Blood pressure augmentation and maternal circulating concentrations of angiogenic factors at delivery in preeclamptic and uncomplicated pregnancies. Am J Obstet Gynecol 2008;199(6):653 e1-10. [ Links ]
8. Purwos unu Y, Sekizawa A, Farina A, et al. Evaluation of physiological alterations of the placenta through analysis of cell -free messenger ribonucleic acid concentrations of angiogenic factors. Am J Obstet Gynecol 2008;198(1):124 e1-7. [ Links ]
9. Lopez - Novoa JM. Soluble endoglin is an accurate predictor and a pathogenic molecule in pre -eclampsia. Nephrol Dial Transplant 2007;22(3):712 -714. [ Links ]
10. Crove tto F, Figueras F, Triunfo S, et al. Added value of angiogenic factors for the prediction of early and late preeclampsia in the first trimester of pregnancy. Fetal Diagn Ther 2014;35(4):258 -266. [ Links ]
11. Gilbe rt JS, Gilbert SA, Arany M, Granger JP. Hypertension produced by placental ischemia in pregnant rats is associated with increased soluble endoglin expression. Hypertension 2009;53(2):399-403. [ Links ]
12. Gilbe rt JS, Babcock SA, Granger JP. Hypertension produced by reduced uterine perfusion in pregnant rats is associated with increased soluble fms -like tyrosine kinase -1 expression. Hypertension 2007;50(6):1142 -1147. [ Links ]
13. Lopez -Novoa JM, Bernabeu C. The physiological role of endoglin in the cardiovascular system. Am J Physiol Heart Circ Physiol 2010;299(4):H959 -974. [ Links ]
14. Venka tesha S, Toporsian M, Lam C, et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat Med 2006;12(6):642 -649. [ Links ]
15. Pijne nborg R, Dixon G, Robertson WB, Brosens I. Trophoblastic invasion of human decidua from 8 to 18 weeks of pregnancy. Placenta 1980;1(1):3-19. [ Links ]
16. Powe CE, Levine RJ, Karumanchi SA. Preeclampsia, a disease of the maternal endothelium: the role of antiangiogenic factors and implications for later cardiovascular disease. Circulation 2011;123(24):2856 -2869. [ Links ]
17. Zhou Y, Fisher SJ, Janatpour M, et al. Human cytotrophoblasts adopt a vascular phenotype as they differentiate. A strategy for successful endovascular invasion? J Clin Invest 1997;99(9):2139 -2151. [ Links ]
18. Lyall F. Priming and remodelling of human placental bed spiral arteries during pregnancy–a review. Placenta 2005;26 Suppl A:S31 -36. [ Links ]
19. Genba cev O, Zhou Y, Ludlow JW, Fisher SJ. Regulation of human placental development by oxygen tension. Science 1997;277(5332):1669 -1672. [ Links ]
20. Al -Gu bory KH, Fowler PA, Garrel C. The roles of cellular reactive oxygen species, oxidative stress and antioxidants in pregnancy outcomes. Int J Biochem Cell Biol 2010;42(10):1634 -1650. [ Links ]
21. Soley manlou N, Jurisica I, Nevo O, et al. Molecular evidence of placental hypoxia in preeclampsia. J Clin Endorinol Metab 2005;90(7):4299-4308. [ Links ]
22. Grang er JP, Alexander BT, Llinas MT, Bennett WA, Khalil RA. Pathophysiology of preeclampsia: linking placental ischemia/hypoxia with microvascular dysfunction. Microcirculation 2002;9(3):147-160. [ Links ]
23. Abitb ol MM, Pirani CL, Ober WB, Driscoll SG, Cohen MW. Production of experimental toxemia in the pregnant dog. Obstet Gynecol 1976;48(5):537-548. [ Links ]
24. Abitb ol MM, Gallo GR, Pirani CL, Ober WB. Production of experimental toxemia in the pregnant rabbit. Am J Obstet Gynecol 1976;124(5):460-470. [ Links ]
25. Loson czy G, Brown G, Venuto RC. Increased peripheral resistance during reduced uterine perfusion pressure hypertension in pregnant rabbits. Am J Med Sci 1992;303(4):233 -240. [ Links ]
26. Combs CA, Katz MA, Kitzmiller JL, Brescia RJ. Experimental preeclampsia produced by chronic constriction of the lower aorta: validation with longitudinal blood pressure measurements in conscious rhesus monkeys. Am J Obstet Gynecol 1993;169(1):215 -223. [ Links ]
27. Hodar i AA. Chronic uterine ischemia and reversible experimental toxemia of pregnancy. Am J Obstet Gynecol 1967;97(5):597-607. [ Links ]
28. Alexa nder BT, Kassab SE, Miller MT, et al. Reduced uterine perfusion pressure duringm pregnancy in the rat is associated with increases in arterial pressure and changes in renal nitric oxide. Hypertension 2001;37(4):1191-1195. [ Links ]
29. Alexa nder BT, Rinewalt AN, Cockrell KL, Massey MB, Bennett WA, Granger JP. Endothelin type a receptor blockade attenuates the hypertension in response to chronic reductions in uterine perfusion pressure. Hypertension 2001;37(2 Pt 2):485 -489. [ Links ]
30. Alexa nder BT, Cockrell K, Cline FD, Llinas MT, Sedeek M, Granger JP. Effect of angiotensin II synthesis blockade on the hypertensive response to chronic reductions in uterine perfusion pressure in pregnant rats. Hypertension 2001;38(3 Pt 2):742 -745. [ Links ]
31. Crews JK, Herrington JN, Granger JP, Khalil RA. Decreased endothelium-dependent vascular relaxation during reduction of uterine perfusion pressure in pregnant rat. Hypertension 2000;35(1 Pt 2):367-372. [ Links ]
32. Grang er JP, LaMarca BB, Cockrell K, et al. Reduced uterine perfusion pressure (RUPP) model for studying cardiovascular -renal dysfunction in response to placental ischemia. Methods Mol Med 2006;122:383-392. [ Links ]
33. Gougo s A, St Jacques S, Greaves A, et al. Identification of distinct epitopes of endoglin, an RGD-containing glycoprotein of endothelial cells, leukemic cells, and syncytiotrophoblasts. Int Immunol 1992;4(1):83 -92. [ Links ]
34. Cheif etz S, Bellon T, Cales C, et al. Endoglin is a component of the transforming growth factor -beta receptor system in human endothelial cells. J Biol Chem 1992;267(27):19027-19030. [ Links ]
35. Topor sian M, Gros R, Kabir MG, et al. A role for endoglin in coupling eNOS activity and regulating vascular tone revealed in hereditary hemorrhagic telangiectasia. Circ Res 2005;96(6):684 -692. [ Links ]
36. Oujo B, Perez -Barriocanal F, Bernabeu C, Lopez -Novoa JM. Membrane and soluble forms of endoglin in preeclampsia. Curr Mol Med 2013;13(8):1345 -1357. [ Links ]
37. Farin a A, Sekizawa A, De Sanctis P, et al. Gene expression in chorionic villous samples at 11 weeks gestation from women destined to develop preeclampsia. Prenat Diagn 2008;28(10):956 -961. [ Links ]
38. Rodes ch F, Simon P, Donner C, Jauniaux E. Oxygen measurements in endometrial and trophoblastic tissues during early pregnancy. Obstet Gynecol 1992;80(2):283 -285. [ Links ]
39. Burto n GJ, Caniggia I. Hypoxia: implications for implantation to delivery-a workshop report. Placenta 2001;22 Suppl A:S63 -65. [ Links ]
40. Gu Y, Lewis DF, Wang Y. Placental productions and expressions of soluble endoglin, soluble fms -like tyrosine kinase receptor -1, and placental growth factor in normal and preeclamptic pregnancies. J Clin Endocrinol Metab 2008;93(1):260 -266. [ Links ]
41. Sanch ez -Elsner T, Botella LM, Velasco B, Langa C, Bernabeu C. Endoglin expression is regulated by transcriptional cooperation between the hypoxia and transforming growth factor -beta pathways. J Biol Chem 2002;277(46):43799-43808. [ Links ]
42. Canig gia I, Winter JL. Adriana and Luisa Castellucci Award lecture 2001. Hypoxia inducible factor -1: oxygen regulation of trophoblast differentiation in normal and pre- eclamptic pregnancies– a review. Placenta 2002;23 Suppl A:S47-57. [ Links ]
43. Yinon Y, Nevo O, Xu J, et al. Severe intrauterine growth restriction pregnancies have increased placental endoglin levels: hypoxic regulation via transforming growth factor-beta 3. Am J Pathol 2008;172(1):77-85. [ Links ]
44. Canig gia I, Mostachfi H, Winter J, et al. Hypoxia -inducible factor-1 mediates the biological effects of oxygen on human trophoblast differentiation through TGFbeta(3). J Clin Invest 2000;105(5):577-587. [ Links ]
45. Tal R , Shaish A, Barshack I, et al. Effects of hypoxia -inducible factor-1alpha overexpression in pregnant mice: possible implications for preeclampsia and intrauterine growth restriction. Am J Pathol 2010;177(6):2950-2962. [ Links ]
46. Chand el NS, McClintock DS, Feliciano CE, et al. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia -inducible factor -1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem 2000;275(33):25130 -25138. [ Links ]
47. Tam T am KB, Lamarca B, Arany M, et al. Role of reactive oxygen species during hypertension in response to chronic antiangiogenic factor (sFlt -1) excess in pregnant rats. Am J Hypertens 2011;24(1):110 -113. [ Links ]
48. Wang Y, Walsh SW. Antioxidant activities and mRNA expression of superoxide dismutase, catalase, and glutathione peroxidase in normal and preeclamptic placentas. J Soc Gynecol Investig 1996;3(4):179 -184. [ Links ]
49. Cudmo re M, Ahmad S, Al -Ani B, et al. Negative regulation of soluble Flt -1 and soluble endoglin release by heme oxygenase -1. Circulation 2007;115(13):1789 -1797. [ Links ]
50. Wakat suki A, Ikenoue N, Okatani Y, Shinohara K, Fukaya T. Lipoprotein particles in preeclampsia: susceptibility to oxidative modification. Obstet Gynecol 2000;96(1):55 -59. [ Links ]
51. Qiu C , Phung TT, Vadachkoria S, Muy -Rivera M, Sanchez SE, Williams MA. Oxidized low -density lipoprotein (Oxidized LDL) and the risk of preeclampsia. Physiol Res 2006;55(5):491 -500. [ Links ]
52. Repa JJ, Mangelsdorf DJ. The role of orphan nuclear receptors in the regulation of cholesterol homeostasis. Annu Rev Cell Dev Biol 2000;16:459 -481. [ Links ]
53. Bourd eau A, Dumont DJ, Letarte M. A murine model of hereditary hemorrhagic telangiectasia. J Clin Invest 1999;104(10):1343 -1351. [ Links ]
54. Mouza t K, Mercier E, Polge A, et al. A common polymorphism in
55. Valbu ena -Diez AC, Blanco FJ, Oujo B, et al. Oxysterol -induced soluble endoglin release and its involvement in hypertension. Circulation 2012;126(22):2612 -2624. [ Links ]
56. Henry -Berger J, Mouzat K, Baron S, et al. Endoglin (CD105) expression is regulated by the liver X receptor alpha (NR1H3) in human trophoblast cell line JAR. Biol Reprod 2008;78(6):968 -975. [ Links ]
57. Hawin kels LJ, Kuiper P, Wiercinska E, Verspaget HW, Liu Z, Pardali E, et al. Matrix metalloproteinase -14 (MT1-MMP)-mediated endoglin shedding inhibits tumor angiogenesis. Cancer Res 2010;70(10):4141 -4150. [ Links ]
58. Kaitu u -Lino TJ, Palmer KR, Whitehead CL, Williams E, Lappas M, Tong S. MMP -14 is expressed in preeclamptic placentas and mediates release of soluble endoglin. Am J Pathol 2012;180(3):888 -894. [ Links ]
59. Kaitu u -Lino TJ, Palmer K, Tuohey L, Ye L, Tong S. MMP -15 is upregulated in preeclampsia, but does not cleave endoglin to produce soluble endoglin. PloS one 2012;7(6):e39864. [ Links ]
60. Kaitu u -Lino TJ, Tuohey L, Ye L, Palmer K, Skubisz M, Tong S. MT -MMPs in pre –eclamptic placenta: relationship to soluble endoglin production. Placenta 2013;34(2):168 -173. [ Links ]
61. Zhou CC, Irani RA, Zhang Y, et al. Angiotensin receptor agonistic autoantibody –mediated tumor necrosis factor -alpha induction contributes to increased soluble endoglin production in preeclampsia. Circulation 2010;121(3):436 -444. [ Links ]
62. Santi banez JF, Letamendia A, Perez -Barriocanal F, et al. Endoglin increases eNOS expression by modulating Smad2 protein levels and Smad2 -dependent TGF beta signaling. J Cell Physiol 2007;210(2):456 -468. [ Links ]
63. Luft FC. Soluble endoglin (sEng) joins the soluble fms -like tyrosine kinase (sFlt) receptor as a pre -eclampsia molecule. Nephrol Dial Transplant 2006;21(11):3052 -3054. [ Links ]
64. Lowe DT. Nitric oxide dysfunction in the pathophysiology of preeclampsia. Nitric Oxide 2000;4(4):441 -458. [ Links ]
65. Shese ly EG, Maeda N, Kim HS, et al. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc Natl Acad Sci USA 1996;93(23):13176 -13181. [ Links ]
66. Grego ry AL, Xu G, Sotov V, Letarte M. Review: the enigmatic role of endoglin in the placenta. Placenta 2014;35 Suppl:S93 -99. [ Links ]
67. Balle rmann BJ. Contribution of the endothelium to the glomerular permselectivity barrier in health and disease. Nephron Physiol 2007;106(2):p19 -25. [ Links ]
68. Craic i IM, Wagner SJ, Weissgerber TL, Grande JP, Garovic VD. Advances in the pathophysiology of pre -eclampsia and related podocyte injury. Kidney Int 2014;86(2):275 -285. [ Links ]
69. Henao DE, Saleem MA. Proteinuria in preeclampsia from a podocyte injury perspective. Curr Hypertens Rep 2013;15(6):600 -605. [ Links ]
70. Rossi E, Sanz -Rodriguez F, Eleno N, et al. Endothelial endoglin is involved in inflammation: role in leukocyte adhesion and transmigration. Blood 2013;121(2):403-415. [ Links ]
71. Sachs N, Sonnenberg A. Cell -matrix adhesion of podocytes in physiology and disease. Nat Rev Nephrol 2013;9(4):200 -210. [ Links ]
72. Canig gia I, Taylor CV, Ritchie JW, Lye SJ, Letarte M. Endoglin regulates trophoblast differentiation along the invasive pathway in human placental villous explants. Endocrinology 1997;138(11):4977-4988. [ Links ]
73. Mo J, Fang SJ, Chen W, Blobe GC. Regulation of ALK -1 signaling by the nuclear receptor LXRbeta. J Biol Chem 2002;277(52):50788-50794. [ Links ]
74. Dong W, Xu QX, Han YH, Yin LR. [Inhibitory effects of soluble endoglin on invasive ability of cultured cytotrophoblasts of first trimester of pregnancy]. Sheng Li Xue Bao 2011;63(3):267 -271. [ Links ]
75. Nakas hima A, Yamanaka -Tatematsu M, Fujita N, et al. Impaired autophagy by soluble endoglin, under physiological hypoxia in early pregnant period, is involved in poor placentation in preeclampsia. Autophagy 2013;9(3):303 -316. [ Links ]
José M. López-Novoa, Ph.D.
Departamento de Fisiología y Farmacología; Edificio Departamental;
Campus Miguel de Unamuno; 37007 Salamanca (Spain)
E-mail: jmlnovoa@usal.es
Acknowledgments
The authors thank Dr. Carmelo Bernabeu, (Centro de Investigaciones Biologicas, CSIC, Madrid, Spain) for his advice with the endoglin projects. Studies from the authors laboratories have been supported by grants from Ministerio de Economia y Competitividad of Spain (SAF2010 -15881 and SAF2013-45784-R), Junta de Castilla y Leon (Grant GR100), Red de Investigacion Cooperativa en Enfermedades Renales (Red -inRen, RD06/0016/0013 and RD12/0021/0032) and Instituto Reina Sofia de Investigacion Nefrologica, Fundacion Renal Inigo Alvarez de Toledo. Red-inRen is an initiative of the Instituto de Port J Nephrol Hypert 2014; 28(3): 185-192 191 Salud Carlos III (ISCIII) of Spain supported by FEDER funds. LP-R is supported by a fellowship from Junta de Castilla y Leon(ORDEN EDU/346/2013).
Conflict of interest statement: None declared
Received for publication: 28/07/2014
Accepted: 30/07/2014


{kind=link}