










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
Print version ISSN 0872-0169
Port J Nephrol Hypert vol.28 no.3 Lisboa Sept. 2014
ORIGINAL ARTICLE
Renal amyloidosis: classification of 102 consecutive cases
Amiloidose renal: classificação de 102 casos consecutivos
Isabel Tavares1,2, Raquel Vaz1,2, Luciana Moreira3,4, Pedro Rodrigues Pereira5, Susana Sampaio1,2, J. Ramon Vizcaino6, Joao Paulo Oliveira2,7, Paulo Pinho Costa3,4, Luisa Lobato4,8
1 Department of Nephrology, Centro Hospitalar de São João. Porto, Portugal.
2 Nephrology and Infectious Diseases Research and Development Group – INEB (I3S), University of Porto. Porto, Portugal.
3 National Institute of Health, INSA. Porto, Portugal.
4 Multidisciplinary Unit for Biomedical Research UMIB, Instituto de Ciências Biomédicas Abel Salazar, University of Porto. Porto, Portugal.
5 Departme nt of Pathology, Centro Hospitalar de São João. Porto, Portugal.
6 Department of Pathology, Hospital de Santo António – Centro Hospitalar do Porto. Porto, Portugal.
7 Department of Genetics, Faculty of Medicine, University of Porto. Porto, Portugal.
8 Department of Nephrology, Hospital de Santo António – Centro Hospitalar do Porto. Porto, Portugal.
ABSTRACT
Amyloidoses are a group of heterogeneous diseases classified according to the nature of their causative amyloid proteins. Commonly, paraffin-embedded tissue is used for the typing of amyloid by immunohistochemistry.
DNA analysis should always be considered if hereditary amyloidosis is suspected. Since the kidneys are one of the organs that are most commonly involved in amyloid deposition in systemic amyloidoses, we screened 102 consecutive cases with biopsy-proven amyloid disease by immunohistochemistry. DNA analysis was performed to confirm a diagnosis of hereditary amyloidosis. Demographic characteristics, underlying disease and clinical data at the time of renal biopsy were obtained by retrospective review of medical records.
The amyloidosis type according to immunohistochemical amyloid protein identification was AA in 60 (58.8%) patients, AL in 21 (20.6%), AFib in four (3.9%), ATTR in two (2.0%), AApoAI in one (1.0%), ALys in one (1.0%) and combined AL and AA in one (1.0%). The type of protein could not be classified in 12 (11.7%) patients: eight (7.8%) because of negative immunohistochemistry and four (3.9%) due to the lack of adequate tissue. DNA analysis confirmed AFib and ATTR cases by the identification of the point mutations FGA p.Glu545Val and TTR p.Met51Val, respectively. Mean age at diagnosis was 53.3 years (49.4 for AA, 63.0 for AL and 53.9 for AFib). Chronic infections were the most frequent disorder associated with AA amyloidosis, mainly tuberculosis, and only one patient had familial AA associated with Muckle-Wells syndrome. Nephrotic syndrome was the most frequent clinical manifestation, independently of the amyloid type.
In our series, AA amyloidosis is still the most frequent type of systemic amyloidoses. Six patients had unequivocal hereditary amyloidosis. Immunohistochemistry did not establish the precursor protein in almost 8% of patients; however, an improvement could be obtained using a wider panel of amyloid antibodies.
Key-Words: Amyloidosis; diagnosis; hereditary; immunohistochemistry;kidney.
RESUMO
As amiloidoses são um grupo heterogéneo de doenças classificadas de acordo com a composição das suas proteínas amiloidogénicas. Frequentemente, os tecidos preservados em parafina são usados para identificação imunohistoquímica. A análise de ADN deve ser sempre considerada se houver suspeita de amiloidose hereditária. Dado que os rins são um dos órgãos mais frequentemente envolvidos nas amiloidoses sistémicas, procedemos à classificação imunohistoquímica de 102 casos consecutivos de doença amiloide confirmada por biópsia renal. A análise de ADN foi realizada para confirmar o diagnóstico de amiloidose hereditária. As características demográficas, doença subjacente e dados clínicos à data da biópsia foram obtidos pela revisão retrospetiva dos registos médicos.
O tipo de amiloidose obtido por identificação imunohistoquímica foi AA em 60 (58,8%) doentes, AL em 21 (20,6%), AFib em quatro (3,9%), ATTR em dois (2,0%), AApoAI em um (2,0%), ALys em um (2,0%), e em um (2,0%) coexistiam os tipos AL e AA. Em 12 (11,7%) não foi identificado o tipo de amiloide: oito (7,8%) por imunohistoquímica negativa e quatro (3,9%) devido a amostra insuficiente. A análise de ADN confirmou os casos AFib e ATTR pela identificação das mutações pontuais FGA p.Glu545Val e TTR p.Met51Val, respetivamente. A média de idade à data do diagnóstico foi 53,3 anos (49,4 para AA, 63,0 para AL e 53,9 para AFib). As infeções crónicas foram a principal causa de amiloidose AA, sobretudo a tuberculose, e foi apenas identificada uma AA familiar associada a síndrome de Muckle-Wells. A síndrome nefrótica foi a manifestação clínica mais frequente, independentemente do tipo de amiloide.
Na nossa série, a amiloidose AA continua a ser a amiloidose sistémica mais frequente. Seis doentes tiveram amiloidose hereditária inequívoca. A imunohistoquímica não identificou a proteína precursora em quase 8% dos doentes; contudo, a utilização de um painel de anticorpos mais alargado poderá melhorar o diagnóstico.
Palavras-chave: Amiloidose; diagnóstico; hereditário; imunohistoquímica; rim.
INTRODUCTION
Amyloidoses comprise a heterogeneous group of diseases that have in common tissue deposits of extracellular fibrillary proteins of similar structure but different chemical composition1-3. Although they have been known since the time of Virchow, in the 19th century4,5, until recently amyloidoses were considered a medical curiosity with only academic interest rather than clinically relevant diseases. However, recent advances in the treatment of systemic amyloidoses have changed this position and, hence, the importance of an early and correct diagnosis of the type of amyloid has gained relevance6-11. The sample to be analysed must be of reasonable quality and quantity. Currently, amyloid deposits are identified on the basis of their apple-green birefringence under polarized light microscopy in Congo red stained histological preparations, the gold standard for amyloid detection, and the presence of rigid, non-branching fibrils 7.5 to 10 nm in diameter, on electron microscopy12. Immunohistochemical identification of the chemical type of amyloid is still the first step in classifying amyloid13. However, it must be performed and interpreted with caution and inconclusive results must be further evaluated using more sophisticated methods available in referral centres9,14. Additional genetic testing should be performed if a hereditary form is suspected after amyloid protein typing9,15.
In cases in which DNA sequencing detects a mutant amyloid precursor, protein analysis is the definitive evidence9. To date, more than 25 different proteins have been recognized as causative agents of amyloid diseases16. The two most common types of systemic amyloidoses, commonly associated with renal involvement, are immunoglobulin-derived amyloidosis, secondary to plasma cell dyscrasia, and serum amyloid A derived amyloidosis (AA), which is typically associated with chronic inflammation. The deposits in immunoglobulin-derived amyloidosis in the vast majority of patients are composed of fragments of monoclonal immunoglobulin light chains (AL), but seldom develop from fragments of heavy chains (AH)17-19. Other rare forms of amyloidoses with renal involvement are those derived from transthyretin (ATTR)20, gelsolin (AGel)21, apolipoprotein A-I (AApoAI)22, fibrinogen A α-chain (AFib)23, lysozyme (ALys)24, apolipoprotein A-II (AApoAII)25, apolipoprotein A-IV (AApoAIV)26, and from the leukocyte chemotactic factor 2 (ALect2)27. This newly described form of amyloidosis is mainly a renal disease from a clinical perspective, although not enough is known yet about ALect2 to draw conclusions about the distribution of amyloid deposits27. A precise epidemiology of amyloidoses is difficult to define as the disease is often undiagnosed or misdiagnosed. Selection bias of data from tertiary centres becomes potentially unrepresentative1,15. In Portugal, ATTR is the most frequent form of hereditary systemic amyloidosis.
Since the identification of the disease as a new entity, in 1939, Hospital Santo António is the reference centre for this disease. Apart from ATTR, there is not a vast knowledge about the type of systemic amyloidoses in Portugal. The aims of this study were: (1) to classify by immunohistochemistry the type of amyloidosis of a northern Portuguese series evaluated outside the referral centre; (2) to identify amyloidogenic variants in the hereditary forms; (3) to determine underlying disorder, clinical and laboratory findings at the time of kidney biopsy.
SUBJECTS AND METHODS
The department of Nephrology of the Centro Hospitalar São João (CHSJ), in collaboration with the department of Renal Pathology of CHSJ, has a registry of native kidney biopsies with amyloid nephropathy diagnosed since 1978. The absence of amyloid type classification led the Nephrology department of CHSJ to start a systematic classification of all cases of amyloid nephropathy by immunohistochemistry. We conducted a retrospective review of 102 consecutive native kidney biopsies, from patients of northern Portugal, performed between May 1978 and September 2013. Five of those patients had one or two additional kidney biopsies. For those patients, only the first diagnostic biopsy was analysed. DNA studies were performed for confirmation of the amyloid type in suspected hereditary amyloidosis based on the protein identified in the deposits.
The study was reviewed and approved by the Health Ethics Commission of CHSJ.
Histology and Immunohistochemistry
All the immunohistochemistry slides were reviewed by a pathologist and a nephrologist with expertise in amyloid nephropathy. Congo red staining was performed on 6 μm thick formalin-fixed paraffinembedded sections, and the presence of amyloid was analysed microscopically under polarized light.
Immunohistochemical staining was performed on 2 μm thick formalin-fixed paraffin-embedded sections of the amyloid containing biopsies, using standard methods and a ready-to-use rabbit/mouse, peroxidase/diaminobenzidine (DAB) detection system (Dako Real Envision). The monoclonal antibodies used were directed against serum amiloid A (Dako), apolipoprotein A-II (Abcam) and transthyretin [provided by one of the authors (PPC)]28; polyclonal antibodies were used for κ-light chain, λ-light chain, fibrinogen A α-chain, transthyretin, apolipoprotein A-I and lysozyme (all from Dako). For light chains and fibrinogen detection, sections were treated with 10 μg/mL proteinase K for 10 minutes at 37°C and 10 minutes at room temperature. Blocking was done with 5% bovine serum albumin/phosphate-buffered saline (BSA/PBS). The sections were incubated with the respective antibody for 2 hours at room temperature, diluted in 1% BSA/PBS, as follows: monoclonal anti-TTR was used directly; polyclonal anti-TTR 1:500; anti-SAA 1:100; anti-kappa 1:1000; anti-lambda 1:2000; anti-Fib 1:800; Anti-Lys 1:300; anti-Apo A-I 1:400; anti-Apo A-II 1:600.
DNA Sequence Analysis
We searched for DNA mutations in the genes coding for the proteins identified in the immunohistochemistry.
DNA was isolated from peripheral blood leukocytes using the Genomic DNA Purification Kit (PureGene, Gentra Systems). The coding regions of the genes encoding transthyretin (exon 2), fibrinogen A α-chain (exon 5), apolipoprotein A-I (exons 1 to 4), apolipoprotein A-II (exon 4), and lysozyme (exon 2) were amplified by polymerase-chain-reaction (PCR) using the primer pairs listed on Table I. PCR products were analysed by agarose gel electrophoresis, purified according to High Pure PCR Purification Kit (Roche) and sequenced with the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems). Sequence results were analysed with ChromasPro and Ridom TraceEdit.
Patients and Data Collection
A total of 102 consecutive patients with different types of amyloidoses were included. For each patient, data was obtained from retrospective review of medical records: date of birth, sex, underlying diseases, main clinical manifestation of renal involvement at the date of kidney biopsy.
The following clinical definitions were used: (1) nephrotic syndrome: nephrotic-range proteinuria (≥3.0 g/d) with hypoalbuminemia (< 3.5 g/d) and peripheral oedema; (2) glomerular filtration rate estimation (GFRe) using 2009 CKD-EPI creatinine equation29; (3) hypertension (HTN) was classified according to 2013 ESC/ESH guidelines30.
Statistics
All statistical analyses were performed in IBM SPSS version 20.0 for Windows. Descriptive statistics of nominal variables consisted on frequencies. Kolmogorov-Smirnov test was used to assess the normality of cardinal variables, and for this test the null hypothesis was rejected when p < 0.05. Despite the small size of some subsamples, means and standard deviations were used, as all the described cardinal variables presented normal distribution, and the use of medians and percentiles would not be more appropriate in such small groups.
RESULTS
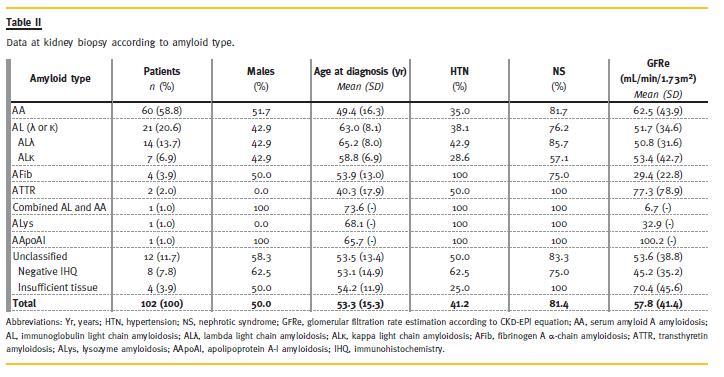
Clinical and laboratory characteristics of the study patients, both overall and according to amyloid type are listed in Table II.Amyloid Type Identification
According to immunohistochemical identification, the most prevalent type of amyloidosis was AA, corresponding to more than half of the patients (58.8%, n = 60), followed by AL in about one fifth (20.6%, n = 21). The ratio AA/AL was 2.9 to 1. The subtype of light chain in AL amyloidosis was λ in two thirds of patients. Two different types of amyloid deposits – light chain λ and serum amyloid A – were present in one patient. Immunostaining disclosed a predominant pattern for light chain λ deposition and small patchy deposits for serum amyloid A at different sites. AFib was the most frequent type of hereditary amyloidosis contributing to 3.9% (n = 4) of the cases. ATTR was identified in two (2.0%) patients, AApoAI in one (1.0%), and ALys in one (1.0%). The type of amyloidosis remained unclassified in 12 (11.7%) patients, mostly due to negative immunohistochemistry.
Hereditary Forms and their Amyloidogenic Variants
Molecular diagnosis of hereditary forms disclosed the point mutations FGA p.Glu545Val in all AFib cases and TTR p.Met51Val in the two cases of ATTR. Sudden death of the ALys positive patient did not allow genetic study. In the AApoAI case pathogenic changes were not detected in the encoding region (exons 2 to 4) and exon-intron respective transitions of the APOA1 gene.
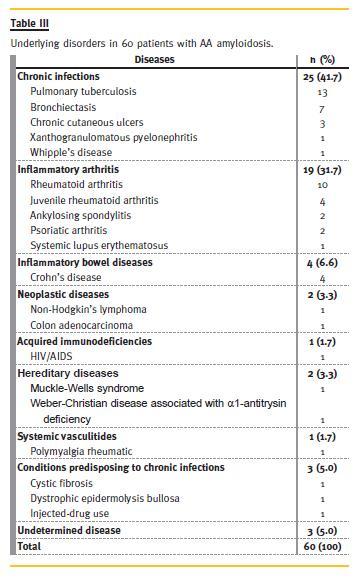
Underlying Disease
In our series, systemic AA amyloidosis patients showed mainly chronic infectious complications of pulmonary tuberculosis (Table III). A familial AA associated to Muckle-Wells syndrome was unequivocal in one patient. AL amyloidosis was related to monoclonal gammopathy of undetermined significance (MGUS) in 10 (47.6%) and multiple myeloma in eight (38.1%) patients, whereas the diagnosis was unknown in three (14.3%). The combined AL and AA amyloidosis patient had multiple myeloma light chain lambda and a previous history of syphilis. One of the four patients with AFib and the AApoAI patient also had MGUS. The ALys patient had rheumatoid arthritis.
Clinical Findings at Kidney Biopsy
The clinical features at kidney biopsy are listed in Table II. The 102 patients included 51 (50.0%) females and 51 (50.0%) males. The mean age at time of kidney biopsy for the entire group was 53.3 ± 14.9 years. AA patients were younger and had a better renal function than AL and AFib. CKD-EPI estimated glomerular filtration rate was 62.5 ± 43.9 mL/min./1.73m2 for AA, 51.7 ± 34.6 mL/min./1.73m2 for AL and 29.4 <± 22.8 mL/min./1.73m2 for AFib patients.
Nephrotic syndrome was the first sign of renal involvement in 83 (81.4%) patients with a similar frequency for all types of amyloidosis. The presence of hypertension was associated with AFib (100% for AFib, 38.1% for AL and 35% for AA).
The mean age of the patients at diagnosis of the underlying disorder was 36 ± 20 years for AA (data available for 53 patients) and 63 ± 9 years for AL (data available for 18 patients). The mean duration between the onset of the underlying disorder and the diagnosis of AA amyloidosis (data available for 53 patients) was 13.9 ± 12.3 years and for AL amyloidosis (data available for 18 patients) was 1.4 ± 1.5 years.
DISCUSSION
Here, we report the immunohistochemical classification, molecular diagnosis and clinical characterization of 102 northern Portuguese patients with kidney biopsy -proven amyloid disease, evaluated outside the referral centre for hereditary amyloidosis.
Diagnosis for amyloid diseases needs histological confirmation31. Immunohistochemistry is still the most frequent technique used in the identification of the amyloid fibril protein. However, it has limitations, mainly related with the low sensitivity of the technique, spectrum of amyloid antibody panel, low quality of the analysed tissue and observer experience15,32-35.
These limitations are particularly evident in the cases of AL and hereditary amyloidosis. Twelve (11.7%) of our cases were unclassified, four (3.9%) due to the lack of adequate tissue and eight (7.8%) because of negative immunohistochemistry. Whenever this occurs, investigation should continue using a wider antibody panel or emerging techniques, such as proteomics, that are currently performed only in highly specialized laboratories14,36-37, which may contribute to amyloid typing in more than 97% of renal amyloidosis cases38.
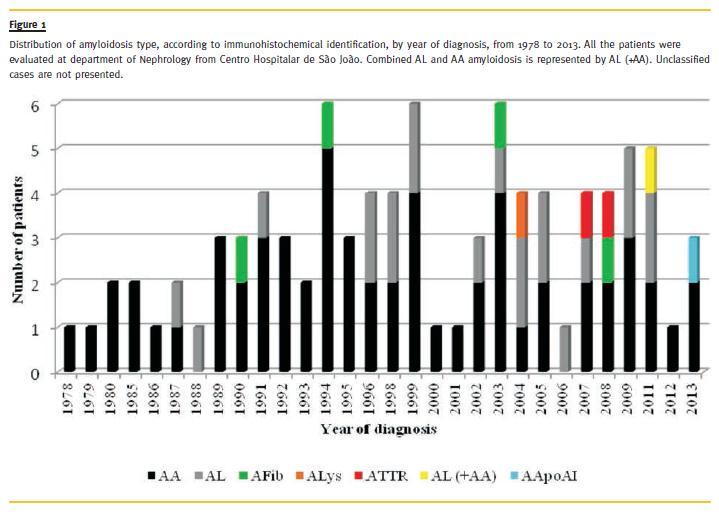
A amyloidosis was the most frequent form of systemic amyloidosis in our series, similarly to what was reported in the largest series of renal amyloidosis in kidney biopsies described by Panizo39. On the other hand, Pinney and colleagues reported an epidemiological study about systemic amyloidosis in England and concluded that systemic AL amyloidosis was the most common type with an estimated minimum incidence of 0.3/100 000 population40. Besides the long period of our study, the prevalence of AA amyloidosis remained stable from 1978 to 2013 (Fig.1), although with recent advances in the treatment of chronic infectious diseases and autoimmune inflammatory processes a decline in the incidence of AA form would be expected39. Nevertheless, our findings agree with results of other series: a recent single-centre study detailing 20 years Florentine experience of AA amyloidosis showed that rheumatoid arthritis contributed to 45% of cases and 67% of patients had some form of renal involvement41. But, among us, chronic infectious disorders are still the most frequent cause of AA amyloidosis, mainly pulmonary tuberculosis, which is according to the fact that Portugal still has one of the highest tuberculosis incidence rates in European Union countries42. One of our AA patients had a Muckle-Wells syndrome, which is a familial autoinflammatory disease, diagnosed when she was 16 years old. Auto-inflammatory diseases are now the most common cause of AA amyloidosis in children, and paediatric nephrologists should be aware that renal amyloidosis is potentially preventable in these conditions43. Clinically significant renal diseases may also arise in young adults with cystic fibrosis44. Our patient had cystic fibrosis since age 5 and AA amyloidosis was diagnosed at 21 years old. Early diagnosis and rapid control of the underlying inflammatory or infectious disease are of the utmost importance to prevent irreversible organ damage45. Our mean duration between the onset of the underlying disease and the diagnosis of AA amyloidosis presented a wide range, nonetheless this was a retrospective observational analysis. During such a long period of time, patient monitoring combined with adequate therapy of underlying disease and periodic search for subclinical amyloid deposits on abdominal fat aspiration, might help early diagnosis and alter the prognosis of the disease45. AA amyloidosis, in our series, affected younger individuals, which may be related to early beginning of most of the underlying conditions, mainly infectious and inflammatory diseases, and therapeutic failures at suppressing inflammation. However, genetic factors may also be involved on this namely SAA genotype46. A lesser degree of renal dysfunction in our patients may be related to an early diagnosis through repeated measurements of microalbuminuria and serum creatinine.
AL amyloidosis was our second most frequent form of systemic amyloidosis and these patients were older than AA patients. These results should be interpreted cautiously since in older and unstable patients with monoclonal plasma cell disorders we may use less invasive diagnostic tests, such as abdominal fat aspiration or minor salivary gland biopsy for amyloid diagnosis as a way to minimize complications related to kidney biopsies.
Unequivocal hereditary amyloidosis contributed to 5.9% of our cases (3.9% AFib and 2.0% ATTR).
DNA analysis is mandatory to confirm the diagnosis but it should always be complementary to other diagnostic techniques that allow unequivocal identification of amyloid protein9. Genetic defects may be associated with amyloidosis either as a mutation in non-amyloid protein, as is the case of familial AA patients, where an inborn error of inflammatory response in the innate immune system plays a permissive role in the development of amyloid47, or as a mutation involving amyloid protein itself, as is the case of AFib23, ATTR48, ALys24 and AApoAI22 patients. AFib is the most common type of hereditary amyloidosis in Europe49. Our four AFib patients were from the same region and had the same amyloidogenic mutation, so haplotyping studies are necessary to conclude if they belong to the same family. Worst renal function at presentation in this small group may not be informative because of late diagnosis, in the absence of family history, due to variations in disease penetrance and progression. High prevalence of hypertension among AFib patients may be secondary to chronic kidney disease, but direct amyloid deposition in vascular walls may also be involved49. ATTR amyloidosis was identified only in two cases, what was expected since this study was performed outside the referral centre for the disease in northern Portugal.
For this reason, our results have no correlation with ATTR prevalence in our country. One of the patients performed kidney biopsy outside the referral centre because she had nephrotic syndrome and at the time of biopsy she denied knowing ATTR family history. The other patient was considered TTR p.Met51Val asymptomatic carrier that presented with nephrotic syndrome and haematuria. Kidney biopsy disclosed IgA nephropathy and medullary amyloid deposition. ALys and AApoAI amyloidosis were diagnosed by immunohistochemistry, without molecular confirmation, so further studies are necessary to confirm amyloidosis type in both cases.
Said and colleagues38 reported the origin and clinicopathologic correlations of 474 recent cases of renal amyloidosis, based on immunohistochemistry and laser microdissection/mass spectrometry. They found immunoglobulin (Ig) amyloidosis in 85.9%; AA in 7%; leukocyte chemotactic factor 2 (LECT2) amyloidosis in 2.7%; AFib in 1.3%; ApoAI, ApoAII or ApoAIV amyloidosis in 0.6%; combined AA /Ig heavy and light chain amyloidosis in 0.2%, and amyloidosis was unclassified in 2.3% patients38. When we compare our results with those, our first conclusion is that we need to raise our spectrum of amyloid antibodies with the inclusion of anti-LECT2, anti-Apo A-IV and anti-Ig heavy chain as a way to reduce unclassified cases.
Improvements to understanding the pathogenesis of systemic amyloidosis, coupled with enhancements on diagnostic techniques, have led to the identification of therapeutic strategies that have already resulted in better outcomes for patients50, so we should perform routine precise identification of the amyloid fibril protein on tissues containing amyloid deposits.
Our results may prove helpful for clinicians in charge of patients with amyloidosis regarding the decision for a more definitive diagnosis of the disease.
Source(s) of support in the form of grants, equipment, drugs, or all of these
This work was supported by a grant from the Portuguese Society of Nephrology and by the Multidisciplinary Unit for Biomedical Research that is funded by grants from the Foundation for Science and Technology (Fcomp-01-0124-FEDER-015893).
References
1. Falk RH, Comenzo RL, Skinner M. The systemic amyloidosis. N Eng J Med 1997; 337(13):898-909. [ Links ]
2. Pepys MB. Pathogenesis, diagnosis and treatment of systemic amyloidosis. Philos Trans R Soc Lond B Biol Sci 2001; 356(1406):203-210. [ Links ]
3. Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Eng J Med 2003; 349(6):583-596. [ Links ]
4. Virchow, R. Zur Cellulosefrage. Virchows Arch Pathol Anat Physiol 1854; 6:416–426. [ Links ]
5. Virchow, R. Über den Gang der amyloiden degeneration. Virchows Arch 1855; 8:364-368. [ Links ]
6. Holmgren G, Ericzon BG, Groth CG, et al. Clinical improvement and amyloid regression after liver transplantation in hereditary transthyretin amyloidosis. Lancet 1993; 341(8853):1113-1116. [ Links ]
7. Sanchorawala V, Skinner M, Quillen K, Finn KT, Doros G, Seldin DC. Long-term outcome of patients with AL amyloidosis treated with high-dose melphalan and stem cell transplantation. Blood 2007; 110(10):3561-3563. [ Links ]
8. Audard V, Matignon M, Weiss L, et al. Successful long-term outcome of the first combined heart and kidney transplant in a patient with systemic AL amyloidosis. Am J Transplant 2009; 9(1):236-240. [ Links ]
9. Picken MM. Amyloidosis – where are we now and where are we heading? Arch Pathol Lab Med 2010; 134(4):545-551. [ Links ]
10. Stangou AJ, Lobato L, Zeldenrust S, et al. Solid organ transplantation for non-TTR hereditary amyloidosis: report from the 1st International Workshop on the Hereditary Renal Amyloidosis. Amyloid 2012; 19(Suppl1):81-84. [ Links ]
11. Pinney JH, Lachmann HJ, Sattianayagam PT, et al. Renal transplantation in systemic amyloidosis-importance of amyloid fibril type and precursor protein abundance. Am J Transplant 2013; 13(2):433-441. [ Links ]
12. Benson MD. Amyloidosis. In: Scriver CR, Beaudet AL, Shy WS et al. Eds. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill 2000: 5345-5378. [ Links ]
13. Murphy CL, Eulitz M, Hmcic R, et al. Chemical typing of amyloid protein contained in formalin-fixed paraffin-embedded biopsy specimens. Am J Clin Pathol 2001; 116(1):135-142. [ Links ]
14. Lavatelli F, Perlman DH, Spencer B, et al. Amyloidogenic and associated proteins in systemic amyloidosis proteome of adipose tissue. Mol Cell Proteomics 2008; 7(8):1570-1583. [ Links ]
15. Lachmann HJ, Booth DR, Booth SE, et al. Misdiagnosis of hereditary amyloidosis as AL (primary) amyloidosis. N Engl J Med 2002; 346(23):1786-1791. [ Links ]
16. Sipe JD, Benson MD, Buxbaum JN, et al. Amyloid fibril protein nomenclature: 2010 recommendations from the nomenclature committee of the International Society of Amyloidosis. Amyloid 2010; 17(3-4):101-104. [ Links ]
17. Mai HL, Sheikh-Hamad D, Herrera GA, Gu X, Tiworg LD. Immunoglobulin heavy chain can be amyloidogenic: morphologic characterization, including immunoelectron microscopy. Am J Surg Pathol 2003; 27(4):541-545. [ Links ]
18. Picken MM. Immunoglobulin light and heavy chain amyloidosis AL/AH: renal pathology and differential diagnosis. Contrib Nephrol 2007; 153:135-155. [ Links ]
19. Sethi S, Theis JD, Leung N, et al. Mass spectrometry-based proteomic diagnosis of renal immunoglobulin heavy chain amyloidosis. Clin J Am Soc Nephrol 2010; 5(12):2180-2187. [ Links ]
20. Lobato L. Familial amyloidotic polyneuropathy: how transthyretin associated amyloidosis involves the kidney. Port J Nephrol Hypert 2008; 22(1):23-30. [ Links ]
21. Maury CP, Alli K, Baumann M. Finnish hereditary amyloidosis. Amino acid sequence homology between the amyloid fibril protein and human plasma gelsoline. FEBS Lett 1990; 260(1):85-87. [ Links ]
22. Nichols WC, Dwulet FE, Liepnieks J, Benson MD. Variant apolipoprotein AI as a major constituent of a human hereditary amyloid. Biochem Biophys Res Commun 1988; 156(2):762-768. [ Links ]
23. Benson MD, Liepnieks J, Uemichi T, Wheeler G, Correa R. Hereditary renal amyloidosis associated with a mutant fibrinogen α-chain. Nat Genet 1993; 3(3):252-255. [ Links ]
24. Pepys MB, Hawkins PN, Booth DR, et al. Human lysozyme gene mutations cause hereditary systemic amyloidosis. Nature 1993; 362(6420):553-557. [ Links ]
25. Benson MD, Liepnieks JJ, Yazaki M, et al. A new human hereditary amyloidosis: the result of a stop-codon mutation in the apolipoprotein AII gene. Genomics 2001; 72(3):272-277. [ Links ]
26. Bergström J, Murphy C, Eulitz M, et al. Codeposition of apolipoprotein A-IV and transthyretin in senile systemic (ATTR) amyloidosis. Biochem Biophys Res Commun 2001; 285(4):903-908. [ Links ]
27. Murphy CL, Wang S, Kestler D, et al. Leukocyte chemotactic factor 2 (LECT2)-associated renal amyloidosis: a case series. Am J Kidney Dis 2010; 56(6):1100-1107. [ Links ]
28. Costa PMP. Amiloidoses transtirretínicas, da biopatologia à terapêutica. Porto University Medical Dissertation 1993; 74-77. [ Links ]
29. Levey AS, Stevens LA, Schmid CH, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med 2009; 150(9):604-612. [ Links ]
30. Mancia G, Fagard R, Narkiewicz K, et al. 2013 ESH/ESC Guidelines for the management of arterial hypertension: The Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). Eur Heart J 2013; 34(28):2159-2219. [ Links ]
31. Rocken C, Schwotzer EB, Linke RP, Sager W. The classification of amyloid deposits in clinicopathological practice. Histopathology 1996; 29(4):325-335. [ Links ]
32. Novak L, Cook WJ, Herrera GA, Sanders PW. AL-amyloidosis is underdiagnosed in renal biopsies. Nephrol Dial Transplant 2004; 19(12):3050-3053. [ Links ]
33. Comenzo RL, Zhou P, Fleisher M, Clark B, Teruya-Feldstein J. Seeking confidence in the diagnosis of systemic AL (Ig light chain) amyloidosis: patients can have both monoclonal gammopathies and hereditary amyloid proteins. Blood 2006; 107(9):3489-3491. [ Links ]
34. Landau H, Comenzo RL, Zhou P, et al. AL amyloidosis in a patient with T60A TTR mutation. Amyloid 2006; 13(Suppl 1):40A. [ Links ]
35. Picken MM, Hazenberg BPC, Obici L. Report from the diagnostic interactive session. In: XI International Symposium on Amyloidosis. Skinner M, Berk JL, Connors LH, Seldin DC, Eds. CRC Press, Boca Raton, FL, 2007:377-382. [ Links ]
36. Solomon A, Murphy CL, Westermark P. Misclassification of amyloidosis is unwarranted. Blood 2006; 108(2):776. [ Links ]
37. Sethi S, Theis JD, Leung N, et al. Mass spectrometry-base proteomic diagnosis of renal immunoglobulin heavy chain amyloidosis. Clin J Am Soc Nephrol 2010; 5(12):2180-2187. [ Links ]
38. Said SM, Sethi S, Valeri AM, et al. Renal amyloidosis: origin and clinicopathologic correlations of 474 recent cases. Clin J Am Soc Nephrol 2013; 8(9):1515-1523. [ Links ]
39. Panizo N, Rivera F, López-Gomez JM, Spanish Registry of Glomerulonephritis. Decreasing incidence of AA amyloidosis in Spain. Eur J Clin Invest 2013; 43(8):767-773. [ Links ]
40. Pinney JH, Smith CJ, Taube JB, et al. Systemic amyloidosis in England: an epidemiological study. Br J Haematol 2013; 161(4):525-532. [ Links ]
41. Cania A, Bergesio F, Curciarello G, et al. The Florence Register of Amyloidosis: 20 years experience in the diagnosis and treatment of the disease in the Florence district area. Amyloid 2011; 18(Supp 1)86-88. [ Links ]
42. Direção-Geral de Saúde [www.portaldasaude.pt]. Lisboa: Direção-Geral da Saúde. Programa Nacional de Luta Contra a Tuberculose (PNT), março de 2010 – Dia mundial da tuberculose. [ Links ]
43. Bilginer Y, Akpolat T, Ozen S. Renal amyloidosis in children. Pediatr Nephrol 2011; 26(8):1215-1227. [ Links ]
44. Yahiaoui Y, Jablonski M, Hubert D, et al. Renal involvement in cystic fibrosis: diseases spectrum and clinical relevance. Clin J Am Soc Nephrol 2009; 4(5):921-928. [ Links ]
45. Obici L, Merlini G. AA Amyloidosis: basic knowledge, unmet needs and future treatments. Swiss Med Wkly 2012; 142:w13580. [ Links ]
46. Nakamura T, Higashi S, Tomada K, Tsukano M, Shono M. Significance of SAA1.3 allele genotype in Japanese patients with amyloidosis secondary to rheumatoid arthritis. Rheumatology (Oxford) 2006; 45(1):43-49. [ Links ]
47. Ryan JG, Kastner DL. Fevers, genes, and innate immunity. Curr Top Microbiol Immun
ol 2008; 321:169-184.
48. Lobato L, Beirão I, Silva M, et al. End-stage renal disease and dialysis in hereditary amyloidosis TTR V30M: presentation, survival and prognostic factors. Amyloid 2004; 11(1): 27-37. [ Links ]
49. Picken MM. Fibrinogen amyloidosis: the clot thickens! Blood 2010; 115(15):2985-2986. [ Links ]
50. Gillmore JD, Hawkins PN. Pathophysiology and treatment of systemic amyloidosis. Nat Rev Nephrol 2013; 9(10):574-586. [ Links ]
Drª Isabel Tavares
Department of Nephrology, Centro Hospitalar de São João, Alameda
Prof. Hernâni Monteiro, 4200-319 Porto, Portugal
E-mail: isabelpts@sapo.pt
Conflict of Interest Statement
None declared.
Acknowledgments
The authors gratefully acknowledge: Dr. Pedro Lacerda from the National Institute of Health, INSA, Porto, for his contribution to the genetic study; Dr. Rui Poínhos from the Faculty of Nutrition and Food Sciences, University of Porto, for his contribution to the statistical analysis; all the colleagues that made this work possible by referring and caring for the patients.
Received for publication: 25/04/2014
Accepted in revised form: 10/07/2014


{kind=link}
{kind=link}
{kind=link}