










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
versão impressa ISSN 0872-0169
Port J Nephrol Hypert vol.28 no.3 Lisboa set. 2014
ORIGINAL ARTICLE
Focal segmental glomerulosclerosis in IgA nephropathy with regard to Oxford classification: Does it matter?
Hamid Nasri1, Muhammed Mubarak2
1 Department of Nephrology, Division of Nephropathology, Isfahan University of Medical Sciences. Isfahan, Iran.
2 Histopathology Department, Sindh Institute of Urology and Transplantation. Karachi, Pakistan.
ABSTRACT
Background: The lesions resembling focal segmental glomerulosclerosis (FSGS) are frequently found concurrently with IgA nephropathy (IgAN). However, there is controversy regarding the significance of this co -existence. In this study, we sought to determine the significance of FSGS-like lesions in a group of IgAN patients, especially with regard to the Oxford classification. Patients and Methods: The FSGS lesions were typed according to the Columbia classification and correlated with clinico -pathological parameters including Oxford classification criteria. Individual lesions suggesting FSGS were also evaluated. IgAN lesions were classified according to the Oxford classification. Results: Of 114 patients, 80 (70.2 %) were male. The mean age of patients was 37.7 ± 13.6 years. Of 35 patients with co-existent IgAN and FSGS, 50% had classic variant (not otherwise specified; NOS), 30% had tip and 20% perihilar variant of FSGS. None of the patients had collapsing or cellular variants. A significantly positive association of FSGS with M (p = 0.001), S (p = 0.001) and T p = 0.001) variables of Oxford classification was found. No significant difference of FSGS, hyalinosis, capsular adhesions and podocytopathy was found between males and females (p > 0.05). However, a significantly positive association of proteinuria (p = 0.037) and renal dysfunction (p = 0.026) with podocytopathy, and of serum creatinine and renal dysfunction with the presence of FSGS (p = 0.027, p = 0.001, respectively) was seen. Conclusion: In conclusion, the concurrence of FSGS -like lesions with IgAN is associated with the established poor clinical and pathological prognostic factors of this disease and the lesions appear to be of prognostic significance. The presence of such lesions should be sought diligently on renal biopsy examination.
Key-Words: Columbia classification focal segmental glomerulosclerosis; IgA nephropathy; Oxford classification; proteinuria; renal failure.
INTRODUCTION
A nephropathy (IgAN) is a very common form of primary glomerulonephritis and occurs worldwide1-5. The reported frequency varies from 2 to 52% of all renal diseases in various studies from different parts of the world6-13. Morphological lesions resembling focal segmental glomerulosclerosis (FSGS) have been found concurrently with some of the primary glomerular diseases such as membranous glomerulopathy or IgAN14-16. Various pathologic processes, which affect the podocytes, lead to one of the histologic subtypes of FSGS15.19. According to the new Oxford classification of IgAN, segmental glomerulosclerosis is defined as a segmental increase in the mesangial matrix with obliteration of capillary lumens and/or adhesion of the tufts with Bowmans capsule (synechiae) 19-22. There are mainly two mechanisms by which segmental sclerotic lesions develop. They may be secondary to an initial segmental proliferative or necrotizing lesion of a glomerular disease or these may be a primary disease as a result of podocyte injury, termed podocytopathy14-18. Segmental glomerulosclerosis (S) is also a morphologic variable included in the Oxford classification developed for IgAN, and is an adverse prognostic indicator for this classification1-5. It is well known that lesions morphologically identical with FSGS may appear in IgAN too15-18. However, there is controversy regarding the significance of this co-existence, and whether this concurrence is the result of two primary renal disorders occurring simultaneously or FSGS represents a final common pathway in the evolution of IgAN18-22. Indeed, some studies suggest that FSGS is secondary to another glomerular injury and/or hypertension in the background of IgAN, or alternatively, they are both due to primary podocyte injury19-23. It was shown that loss of podocytes with denudation of the underlying glomerular basement membrane (GBM) is followed by an adhesion to Bowmans capsule and, thus, the formation of a sclerotic lesion14,15. In various cases of primary FSGS, there is evidence for a circulating toxin that damages the podocytes, while in other settings the podocyte damage may be secondary to haemodynamic changes as a response to loss of functioning nephrons14-18. In the Oxford classification of IgAN, the presence of S lesion was one of the four morphologic variables that were found to predict an adverse outcome. The other variables consisted of mesangial hypercellularity (M), endocapillary hypercellularity (E), and tubular atrophy/interstitial fibrosis (T)2-5, 17-26. In this study, we sought to study the significance of sclerotic lesions resembling idiopathic FSGS in a group of IgAN patients. We secondly aimed to find the association of FSGS lesions with morphologic variables of Oxford classification in our patients.
PATIENTS AND METHODS
After publication of the Oxford classification of IgAN, in 200925, 26, we applied it in this study for the classification of IgAN.
Definition of IgAN
The pathologic diagnosis of IgAN requires the demonstration of IgA -dominant mesangial or mesangial-capillary immune deposits through immunofluorescence (IF) microscopy. The immune deposits were semi-quantitatively assessed as 0 to 3+ positive bright. The definition of IgAN needs the presence of diffuse and global IgA deposits that are more than trace in positivity26-29. The renal biopsies from July 2009 to July 2012 were sent to a reference laboratory.
None of the patients was treated before the biopsy. Biopsies with less than 8 glomeruli were excluded from the study. None of the patients was diagnosed as primary IgAN, having history of collagen vascular diseases, diabetes and liver cirrhosis, based on a questionnaire filled at the time of biopsy admission, laboratory data in patients records and a brief history provided by referee physicians at the time of biopsy admission. The study was approved by the institutional ethical review committee and written informed consent was obtained from all patients at the time of biopsy. Strict adherence to Declaration of Helsinki was observed during the study.
Histologic data
All kidney biopsies were prepared for light and direct IF microscopy. Tissue was fixed in 10% formalin for histologic sectioning. Each renal biopsy was prepared by cutting paraffin blocks into 3 μm sections and staining 2 slides with periodic acid -Schiff (PAS), 2 slides for haematoxylin and eosin (HE), 1 slide for Jones methenamine silver (JMS) and 1 slide for trichrome. Each slide contained 2-3 sections. Materials used for IF were snap -frozen in liquid nitrogen.
Sections (6μm in thickness) were stained for IF study with fluorescein isothiocyanate (FITC) –conjugated antibodies specific for human IgG, IgM, IgA, C1q, C3 and fibrin (DAKO, Produktionsvej 42, DK -2600 Glostrup, Denmark)26-29. IF slides were reported in a scale of 0 to 3+ intensity. IF review was performed without information on patients data and before reviewing the slides for light microscopy. After IF diagnosis of IgAN, histopathology glass slides were reviewed to define the morphologic variables, which were applied in Oxford -MEST classification method.
All slides were reported by a single nephropathologist (HN), thus ensuring consistency of the results. Tissue for electron microscopy (EM) was saved for future study. EM analysis was not performed in this study.
Definitions of morphologic variables of IgA nephropathy; MEST (Oxford classification)
Total number of glomeruli and the number of glomeruli with global sclerosis were recorded for each biopsy. The presence of mesangial hypercellularity (M), endocapillary proliferation (E ), segmental glomerulosclerosis (S) and the proportion of tubular atrophy and interstitial fibrosis; IF/TA (T ) were estimated as published for Oxford–MEST classification25,26.
Definitions of Variants of FSGS
These were adapted from the Columbia classification of FSGS with some modifications as proposed by El Karoui et al.17, 22. Briefly, the presence of mesangial sclerosis accompanied by podocyte hyperplasia/hypertrophy and intracapillary hyalinosis was assumed as the presence of classic variant of FSGS (not otherwise specified, NOS)14-23, as illustrated in Fig. 1. Note that this definition of FSGS is not synonymous with the S variable of Oxford classification, as described in detail in the figure legend. For the cellular variant also the presence of podocyte alterations was considered mandatory for the diagnosis in the context of IgAN. For other three variants of FSGS (perihilar, collapsing and tip variants), the definitions of Columbia classification for FSGS were applied directly17, 22. Also, we studied the presence of individual histological features suggesting FSGS, such as intracapillary hyalinosis, tuft-capsule adhesion (synechiae), presence of podocytic hyperplasia/hypertrophy and/or podocytic capping14-23 separately from FSGS variants and correlated these with various clinicopathological parameters.
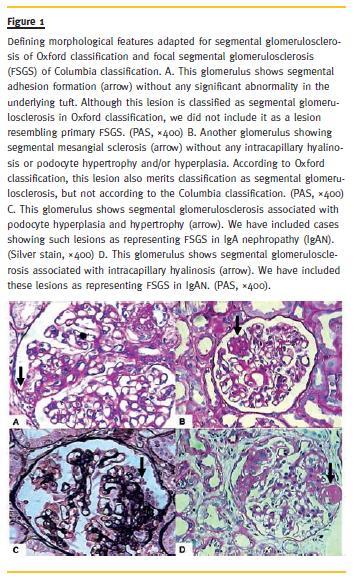
Clinical Studies and Laboratory Data
The medical records of patients were reviewed to obtain various demographic, clinical and laboratory information at the time of biopsy. Data gathered at the time of biopsy included gender, age, serum creatinine and proteinuria (based on a 24-hour urine collection). Renal dysfunction at the time of biopsy was defined as serum creatinine > 1.2 mg/dl in females and > 1.4 mg/dl in males.
Statistical analysis
Results for categorical variables were expressed as numerical values and percentages. Continuous variables were expressed as mean or median values and standard deviations, and statistical significance of the differences between groups was calculated using independent -samples T test, Mann –Whitney U test and Chi -square tests depending on the nature of variables. A computer program (SPSS version 16.0, Chicago, IL, USA) was used for statistical analysis. P < 0.05 was considered statistically significant.
RESULTS
This is an observational study, conducted on IgAN patients. A total of 114 biopsies were enrolled to the study. In this survey of 114 patients, 80 (70.2 %) were male. The mean age of all patients was 37.7 ± 13.6 years (for males, 39 ± 14.3 years and for females, 35 ±11.7 years). The main demographic, clinical and laboratory features of this cohort are summarized in Table 1. The mean value of proteinuria was 1.74 ± 1.32 g/day (median = 1.5 g/day). The mean of serum creatinine was 1.6 ± 1.5 mg/dL (median = 1.2 mg/dL). Renal insufficiency at the time of biopsy was quite common, found in 37.7% of cases. The majority of cases (90.6%) occurred in males.
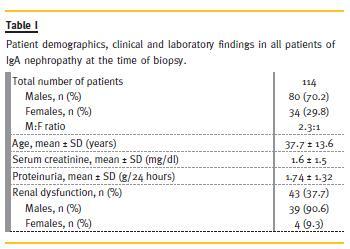
The mean number of total glomeruli in all renal biopsies was 14.8 ± 7.2. The mean number of globally sclerosed glomeruli was 2.4 ± 2.9 (median = 1). The segmental glomerulosclerosis (S) as a morphologic variable of MEST classification was present in 72 (63.2%) patients, but the segmental sclerotic lesion in favour of FSGS was found in 35 (30.7%) patients.
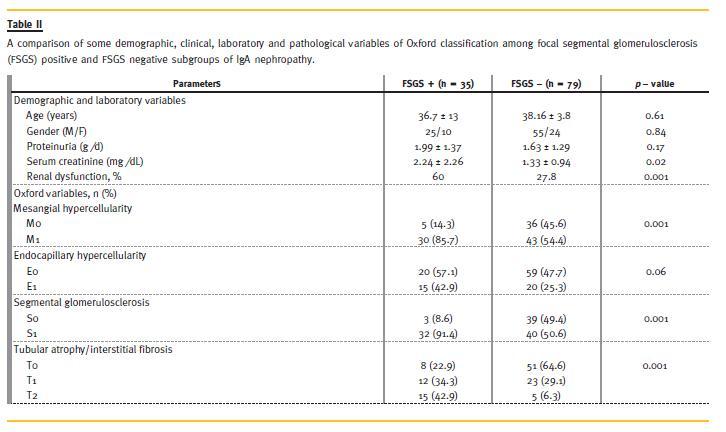
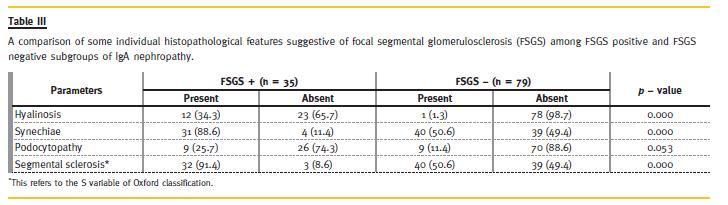
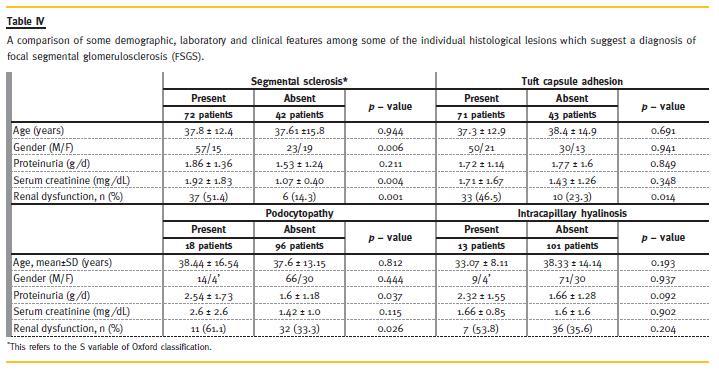
Of 35 patients with co-existence of IgAN and FSGS, 50% were classic variant (not otherwise specified; NOS), 30% had tip variant and 20% the perihilar variant of FSGS. None of the patients had collapsing or cellular variants of FSGS. A correlation of various demographic, clinical, laboratory and the MEST variables with the presence of FSGS is presented in Table 2. Regarding individual histological lesions that suggest FSGS, segmental glomerulosclerosis was found in 72 (63.2%) patients, synechiae formation in 71 (63.3%) patients, and some form of podocytopathy in 18 (15.8%) patients. We also found intracapillary hyalinosis in 13 (11.4 %) patients. The association of these individual lesions of segmental glomerulosclerosis, intracapillary hyalinosis, tuft-capsule adhesion and podocytopathy with FSGS is summarized in Table 3. All of these except podocytopathy correlated strongly with the presence of FSGS (p = 0.001). Finally, a correlation of these four features with some demographic, clinical and laboratory features is shown in Table 4. In this study, no significant difference of FSGS, intracapillary hyalinosis, tuft-capsule adhesion and podocytopathy was found between males and females (p > 0.05). However, a significantly positive association of podocytopathy with proteinuria (p = 0.037) and renal dysfunction (p = 0.026), and of serum creatinine and renal dysfunction with the presence of FSGS (p = 0.027, p = 0.001, respectively) was seen. In this study also, a significant positive association of FSGS with M (p = 0.001), S (p = 0.001) and T (p = 0.001) variables of Oxford classification was also found. No significant association of FSGS with E variable of Oxford classification was seen (p = 0.061).
Amyloid Type Identification
According to immunohistochemical identification, the most prevalent type of amyloidosis was AA, corresponding to more than half of the patients (58.8%, n = 60), followed by AL in about one fifth (20.6%, n = 21). The ratio AA/AL was 2.9 to 1. The subtype of light chain in AL amyloidosis was λ in two thirds of patients. Two different types of amyloid deposits – light chain λ and serum amyloid A – were present in one patient. Immunostaining disclosed a predominant pattern for light chain λ deposition and small patchy deposits for serum amyloid A at different sites. AFib was the most frequent type of hereditary amyloidosis contributing to 3.9% (n = 4) of the cases. ATTR was identified in two (2.0%) patients, AApoAI in one (1.0%), and ALys in one (1.0%). The type of amyloidosis remained unclassified in 12 (11.7%) patients, mostly due to negative immunohistochemistry.
Hereditary Forms and their Amyloidogenic Variants
Molecular diagnosis of hereditary forms disclosed the point mutations FGA p.Glu545Val in all AFib cases and TTR p.Met51Val in the two cases of ATTR. Sudden death of the ALys positive patient did not allow genetic study. In the AApoAI case pathogenic changes were not detected in the encoding region (exons 2 to 4) and exon-intron respective transitions of the APOA1 gene.
Underlying Disease
In our series, systemic AA amyloidosis patients showed mainly chronic infectious complications of pulmonary tuberculosis (Table 3). A familial AA associated to Muckle-Wells syndrome was unequivocal in one patient. AL amyloidosis was related to monoclonal gammopathy of undetermined significance (MGUS) in 10 (47.6%) and multiple myeloma in eight (38.1%) patients, whereas the diagnosis was unknown in three (14.3%). The combined AL and AA amyloidosis patient had multiple myeloma light chain lambda and a previous history of syphilis. One of the four patients with AFib and the AApoAI patient also had MGUS. The ALys patient had rheumatoid arthritis.
Clinical Findings at Kidney Biopsy
The clinical features at kidney biopsy are listed in Table 2. The 102 patients included 51 (50.0%) females and 51 (50.0%) males. The mean age at time of kidney biopsy for the entire group was 53.3 ± 14.9 years. AA patients were younger and had a better renal function than AL and AFib. CKD-EPI estimated glomerular filtration rate was 62.5 ± 43.9 mL/min./1.73m2 for AA, 51.7 ± 34.6 mL/min./1.73m2 for AL and 29.4 ± 22.8 mL/min./1.73m2 for AFib patients.
Nephrotic syndrome was the first sign of renal involvement in 83 (81.4%) patients with a similar frequency for all types of amyloidosis. The presence of hypertension was associated with AFib (100% for AFib, 38.1% for AL and 35% for AA).
The mean age of the patients at diagnosis of the underlying disorder was 36 ± 20 years for AA (data available for 53 patients) and 63 ± 9 years for AL (data available for 18 patients). The mean duration between the onset of the underlying disorder and the diagnosis of AA amyloidosis (data available for 53 patients) was 13.9 ± 12.3 years and for AL amyloidosis (data available for 18 patients) was 1.4 ± 1.5 years.
DISCUSSION
Focal segmental glomerulosclerosis is one of the common histopathological patterns underlying idiopathic nephrotic syndrome (INS), which initially affects a part of some but not all glomeruli associated with the tubulointerstitial and vascular damage of the kidney15-18. It is not a specific disease entity but rather represents a descriptive morphological diagnosis. The clinical diversity and varied histologic features of FSGS contribute to the complex nature of the lesion15-23. Lesions morphologically identical to idiopathic FSGS have been found in association with other primary glomerulopathies, such as membranous nephropathy and IgAN. Some authors have suggested that FSGS serves as a final common pathway in the evolution of other primary glomerular diseases. Still others think that IgAN may be better considered as a variant of FSGS22. The co-existence of IgAN with FSGS has been found to have an adverse effect on the outcome of IgAN30-34. Histologically, FSGS -like IgAN has at most a mild increase in mesangial hypercelluarity and can only be distinguished from primary FSGS on the basis of diffuse mesangial IgA deposits on IF. In the case of co-existence of IgAN and FSGS, some other morphologic variables like podocytopathy (podocytic cobblestoning or podocytic capping) or intracapillary hyalinosis could suggest the presence of FSGS in addition to IgAN16-23. The lesions of FSGS may occur in pure form or be superimposed by other glomerular lesions, such as segmental necrosis, and/or extracapillary proliferation.
Patients with pure FSGS have been shown to have relatively poor survival even without other superimposed glomerular lesions19-23. However, there is scant data in published literature regarding the significance of FSGS-like lesions in the setting of IgAN.
This study is an attempt to determine the significance of FSGS -like lesions in a cohort of IgAN patients of racially uniform population of Iran. Of four morphologic variables of Oxford classification, three (M, S and T) had significantly positive association with the presence of FSGS (p = 0.001 each), while E variable had also a partially positive association (p = 0.06). Regarding individual lesions, the lesions of podocytopathy showed significant association with all four morphological variables (MEST) of Oxford classification. Furthermore, intracapillary hyalinosis had significantly positive association with T variable of IgAN classification. Regarding clinical and laboratory parameters, there was no significant difference of age, gender and proteinuria between FSGS+ and FSGS - subgroups of IgAN. Serum creatinine at presentation was significantly higher in the FSGS+ group and renal dysfunction was also significantly more common in the former subgroup as compared with the FSGS - group. We found a significantly positive association of proteinuria and renal dysfunction with podocytopathy.
A few investigators have previously attempted to determine the prognostic value of lesions of FSGS in IgAN patients. To find the significance of FSGS in mild IgAN, renal biopsies performed, between 1996 and 2005, in adults with a diagnosis of IgAN were reviewed by Weber et al.16. A total of seventy-five patients, 26 (35%) with IgAN and FSGS (FSGS+ group) and 49 (65%) with IgAN without FSGS (FSGS− group) were included in the study. At the time of the renal biopsy, the FSGS+ group had a lower estimated glomerular filtration rate (eGFR), lower serum albumin, higher mean arterial pressure and greater protein excretion than the FSGS− group. On histology, the FSGS+ group had a higher percentage of totally sclerosed glomeruli and 31% of FSGS+ biopsies had ≥ 25% IFTA, while this was not observed in the FSGS−group. They concluded that, FSGS lesion and associated clinical and pathologic findings in patients with mild IgAN are associated with a worse renal outcome16.
In the studies conducted by El Karoui et al. on a cohort of 128 adult patients with IgAN, some form of lesion consistent with FSGS, notably hyalinosis and collapsing glomerulopathy (CG) was found in 101 of their patients22. Those with FSGS had significantly worse renal survival at 80 months than those without. Comparison of pure forms of FSGS (excluding CG) with cases of FSGS having other glomerular lesions (mesangial hyperplasia, endocapillary hypercellularity, glomerular necroses, extracapillary proliferation) revealed that those with FSGS and other superimposed lesions did significantly worse than cases of pure FSGS22.
Our findings broadly support the findings of the previous studies but are not entirely concordant with those reported by El Karoui et al.22. In our study of 112 biopsies of IgAN, 35 patients had lesions typical of FSGS (versus 101/128 in the study by El Karoui et al.). Also none of our biopsies had lesions in favour of collapsing or cellular variants of FSGS. Moreover, intracapillary hyalinosis and podocytopathy were found in 11.4% and 15.5% of our biopsies, respectively.
El Karoui et al. concluded that, the majority of cases of IgAN can be interpreted as representing one or another variant of FSGS22. They also concluded that interpreting IgAN in terms of FSGS emphasizes the role that podocyte lesions may play in the pathogenesis and progression of this disease22.
Indeed, a remarkably high number of their patients, 101 of the total 128, had lesions termed consistent with FSGS, classified by this group as hyalinosis and CG22, 23. These differences may be due to variations in the presentation of IgAN around the world, possibly due to ethnic factors1-6. It is possible that, there are at least three ways by which segmental glomerulosclerosis may occur in the glomeruli in IgAN. First, by post -inflammatory scarring; second, due to compensatory haemodynamic changes following nephron reduction, and finally by primary podocyte damage, perhaps due to mediators released from mesangial cells14-22, 24-36.
We believe that it is possible to distinguish objectively between the S variable of Oxford classification and the FSGS of the Columbia classification. Lesions of podocyte hypertrophy/hyperplasia and intracapillary hyalinosis favour the later over former and these should be looked for carefully when examining renal biopsies from patients with IgAN with segmental glomerular sclerosing lesions. We hypothesize that S variable of Oxford classification represents an early stage in the progression of glomerular lesions in IgAN and the FSGS lesion, the more advanced stage, which finally culminates in diffuse chronic sclerosing glomerulonephritis (GN) and end-stage renal disease (ESRD). We believe that the lesions of FSGS, as defined herein and Columbia classification, represent the morphological markers of progression in IgAN.
We are of the view that the lesions of FSGS do not represent idiopathic FSGS superimposed on IgAN as two separate but concurrent diseases, rather these represent a morphological indicators of impending nephron loss and development of progressive renal failure.
Our study has some limitations too. These include the origin of the study from a single centre in Iran, its retrospective nature and the lack of clinical outcome data. However, we are following this cohort of patients actively and we plan to study the correlation of the MEST variables and FSGS lesions with clinical outcome in the near future. On the other hand, there are many strong points in our study.
These include a racially homogeneous study population, a fair number of patients in the study, no use of pre -biopsy immunosuppressive therapy and the reporting of all biopsies by a single nephropathologist, thus ensuring uniformity of the diagnostic criteria.
In conclusion, our findings firstly corroborate the previous studies regarding the prognostic significance of the presence of FSGS lesions in IgAN.
Secondly, to the best of our knowledge, this represents the first report which studies the association of FSGS of Columbia classification with the MEST variables of Oxford classification. Our findings indicate that the presence of FSGS-like lesions in the background of IgAN is associated with the established poor clinical and pathological prognostic factors of this disease and the lesions appear to be of prognostic significance. The presence of such lesions should be sought diligently on renal biopsy examination.
References
1. Julian BA, Waldo FB, Rifai A, Mestecky J. IgA nephropathy, the most common glomerulonephritis worldwide: A neglected disease in the United States? Am J Med 1988; 84(1):129-132. [ Links ]
2 Nasri H, Mortazavi M, Ghorbani A, et al. Oxford -MEST classification in IgA nephropathy patients: A report from Iran. J Nephropathology 2012; 1(1):31-42. [ Links ]
3 Nasri H. Comment on: Clinical, histopathological and immunofluorescent findings of IgA nephropathy. Iran J Immunol 2012; 9(4):266-267. [ Links ]
4 Mubarak M. Oxford classification of IgA nephropathy: Broadening the scope of the classification. J Nephropathol 2012; 1(1): 13-16. [ Links ]
5 Grcevska L, Ristovska V, Nikolov V, Petrusevska G, Milovanceva-Popovska M, Polenakovic M. The Oxford classification of IgA nephropathy: Single centre experience. Prilozi 2010; 31(2):7-16. [ Links ]
6 Donadio JV, Bergstralh EJ, Grande JP, Rademcher DM. Proteinuria patterns and their association with subsequent end-stage renal disease in IgA nephropathy. Nephrol Dial Transplant 2002; 17(7):1197 -1203. [ Links ]
7 Mubarak M. The prevalence of IgA nephropathy in Pakistan: only a tip of the iceberg. J Pak Med Assoc 2009;59(10):733. [ Links ]
8 Kazi JI, Mubarak M. IgA nephropathy. J Coll Physicians Surg Pak 2010;20(12):779 -780. [ Links ]
9 Seedat YK, Nathoo BC, Parag KB, Naiker IP, Ramsaroop R. IgA nephropathy in blacks and Indians of Natal. Nephron 1988;50(2):137 -141. [ Links ]
10 Le W, Zeng CH, Liu Z, et al. Validation of the Oxford classification of IgA nephropathy for pediatric patients from China. BMC Nephrol 2012;13:158. [ Links ]
11 Koyama A, Igarashi M, Kobayashi M. Natural history and risk factors for immunoglobulin A nephropathy in Japan. Research Group on Progressive Renal Diseases. Am J Kidney Dis 1997; 29(4): 526-532. [ Links ]
12 Haas M. IgA nephropathy histologically resembling focal -segmental glomerulosclerosis: a clinicopathologic study of 18 cases. Am J Kidney Dis 1996; 28(3):365 -371. [ Links ]
13 Mittal N, Joshi K, Rane S, Nada R, Sakhuja V. Primary IgA nephropathy in north India: is it different? Postgrad Med J 2012; 88(1035):15 -20. [ Links ]
14 Shakeel S, Mubarak M, Kazi J, Jafry N, Ahmed E. Frequency and clinicopathological characteristics of variants of primary focal segmental glomerulosclerosis in adults presenting with nephrotic syndrome. J Nephropathol 2013; 2(1): 28 -35. [ Links ]
15 Dumoulin A, Hill GS, Montseny JJ, Meyrier A. Clinical and morphological prognostic factors in membranous nephropathy: significance of focal segmental glomerulosclerosis. Am J Kidney Dis 2003; 41(1):38-48. [ Links ]
16 Weber CL, Rose CL, Magil AB. Focal segmental glomerulosclerosis in mild IgA nephropathy: a clinical -pathologic study. Nephrol Dial Transplant 2009; 24(2):483-488. [ Links ]
17 DAgati VD, Fogo AB, Bruijn JA, Jennette JC. Pathologic classification of focal segmental glomerulosclerosis: a working proposal. Am J Kidney Dis 2004; 43(2):368-382. [ Links ]
18 Cook HT. Focal segmental glomerulosclerosis in IgA nephropathy: a result of primary podocyte injury? Kidney Int 2011; 79(6):581-583. [ Links ]
19 Ibrahim Seif E, Abdel -Salam Ibrahim E, Galal Elhefnawy N, Ibrahim Salman M. Histological patterns of idiopathic steroid resistant nephrotic syndrome in Egyptian children: A single centre study. J Nephropathol 2013; 2(1): 53 -60. [ Links ]
20 -Mubarak M. Collapsing focal segmental glomerulosclerosis: increasing the awareness. J Nephropathol 2012; 1(2):77 -80. [ Links ]
21 Mohammadi Torbati P. Focal segmental glomerulosclerosis; collapsing variant. J Nephropathol 2012; 1(2):87-90. [ Links ]
22 El Karoui K, Hill GS, Karras A, et al. Focal segmental glomerulosclerosis plays a major role in the progression of IgA nephropathy. II. Light microscopic and clinical studies. Kidney Int 2011; 79(6):643-654. [ Links ]
23 Hill GS, Karoui KE, Karras A, et al. Focal segmental glomerulosclerosis plays a major role in the progression of IgA nephropathy. I. Immunohistochemical studies. Kidney Int 2011; 79(6):635 -642. [ Links ]
24 Coppo R, Troyanov S, Camilla R, el al. The Oxford IgA nephropathy clinicopathological classification is valid for children as well as adults. Kidney Int 2010; 77(10):921 -927. [ Links ]
25 Cattran DC, Coppo R, Cook HT, et al. The Oxford classification of IgA nephropathy: rationale, clinicopathological correlations, and classification. Kidney Int 2009;76(5):534- 545. [ Links ]
26 Roberts IS, Cook HT, Troyanov S, et al. The Oxford classification of IgA nephropathy: pathology definitions, correlations, and reproducibility. Kidney Int 2009; 76(5): 546 -556. [ Links ]
27 Mubarak M, Kazi JI, Kulsoom U, Ishaque M. Detection of immunoglobulins and complement components in formalin fixed and paraffin embedded renal biopsy material by immunoflourescence technique. J Nephropathol 2012; 1(2): 91 -100. [ Links ]
28 Bellur SS, Troyanov S, Cook HT, Roberts IS. Immunostaining findings in IgA nephropathy: correlation with histology and clinical outcome in the Oxford classification patient cohort. Nephrol Dial Transplant 2011;26(8):2533 -2536. [ Links ]
29 Mubarak M. Immunostaining findings in IgA nephropathy: correlation with histology and clinical outcome in the Oxford Classification patient cohort. Nephrol Dial Transplant 2012; 27(7):2998 -2999. [ Links ]
30 Fogo AB, Alpers CE, DAgati VD. FSGS lesions in IgA nephropathy. Kidney Int 2011;80(3):319. [ Links ]
31 Bartosik LP, Lajoie G, Sugar L, Cattran DC. Predicting progression in IgA nephropathy. Am J Kidney Dis 2001; 38(4):728-735. [ Links ]
32 DAmico G. Natural history of idiopathic IgA nephropathy and factors predictive of disease outcome. Semin Nephrol 2004;24(3):179 -196. [ Links ]
33 Hill GS, Nochy D, El Karoui K. Comments on the Oxford classification of IgA nephropathy. Kidney Int 2009;76(11):1207. [ Links ]
34 Yamamoto R, Imai E. A novel classification for IgA nephropathy. Kidney Int 2009;76(5):477-480. [ Links ]
35 Nasri H, Sajjadieh S, Mardani S, e al. Correlation of immunostaining findings with demographic data and variables of Oxford classification in IgA nephropathy. J Nephropathol 2013; 2(3): 190-195.
36 Mubarak M. Significance of immunohistochemical findings in Oxford classification of IgA nephropathy: The need for more validation studies. J Nephropathol 2013; 2(3): 210 -213. [ Links ]
Prof. Dr. Muhammed Mubarak
Histopathology Department, Sindh Institute of Urology and Transplantation, Karachi-74200, Pakistan,
E-mail: drmubaraksiut@yahoo.com
Conflict of interest statement: None declared.
Funding/Support: A part of this study was funded by Dr. Baradaran
Laboratory, Isfahan.
Received for publication: 10/05/2014
Accepted in revised form: 13/08/2014


{kind=link}
{kind=link}
{kind=link}