










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
versão impressa ISSN 0872-0169
Port J Nephrol Hypert vol.31 no.4 Lisboa dez. 2017
MINI REVIEW
IgG4-tubulointerstitial nephritis: a literature review following 5 cases in a single centre
Joana Rocha1*, Amy Kang2, Nikki Wong2, Joseph Westaby3, Candice Roufosse3, Terence Cook3
1 Nephrology Department, CHTMAD, Vila Real, Portugal
2 Imperial College Renal and Transplant Centre, Hammersmith Hospital, London, United Kingdom
3 Department of Cellular Pathology, Hammersmith Hospital, London, United Kingdom
ABSTRACT
Immunoglobulin G4‑related disease is a recently described systemic fibro‑inflammatory disease, characterized by an infiltration of abundant IgG4+ plasma cells and lymphocytes, leading to tumour‑like swellings of the involved organs, and variable degrees of fibrosis. Organ involvement can occur metachronously, and as IgG4 expressionin other tissues is less specific, renal biopsy often plays a critical role in diagnosis leading to prompt treatment, which can prevent renal damage and other organ failure. Tubulointerstitial nephritis is the most common renal feature. IgG4‑renal involvement is challenging to diagnose and remains under‑recognized, particularly in patients with single‑organ involvement.
The authors describe the only 5 cases of IgG4‑RD with kidney involvement identified in the last 10 years in a tertiary referral nephrology centre that serves a population of 3 million people.
These cases underline the importance of an early diagnosis of IgG4‑related disease, since early treatment is usually effective. They also corroborate that clinical and histopathological features are heterogeneous and that complement may play a role in its pathogenesis.
Key‑words: IgG4, tubulo‑interstitial nephritis
INTRODUCTION
Immunoglobulin G4‑related disease (IgG4‑RD) is a recently described systemic fibro‑inflammatory disease, characterized by an infiltration of abundant IgG4+ plasma cells and lymphocytes, leading to tumour‑likeswellings of the involved organs, and variable degreesof fibrosis. Organ involvement can occur metachronouslyand, as IgG4 expression in other tissues is lessspecific, renal biopsy (RB) often plays a critical role indiagnosis and prompting treatment, which can preventrenal damage and other organ failure. Tubulointerstitialnephritis (TIN) is the most common renal feature andis the focus of this brief review.
This is a case series report describing the only 5 cases of IgG4‑RD with kidney involvement identified in the last 10 years in a tertiary referral nephrology centre that serves a population of 3 million people. It was the starting point for a literature review regarding recognition and management of this newly described entity.
CASE REPORTS:
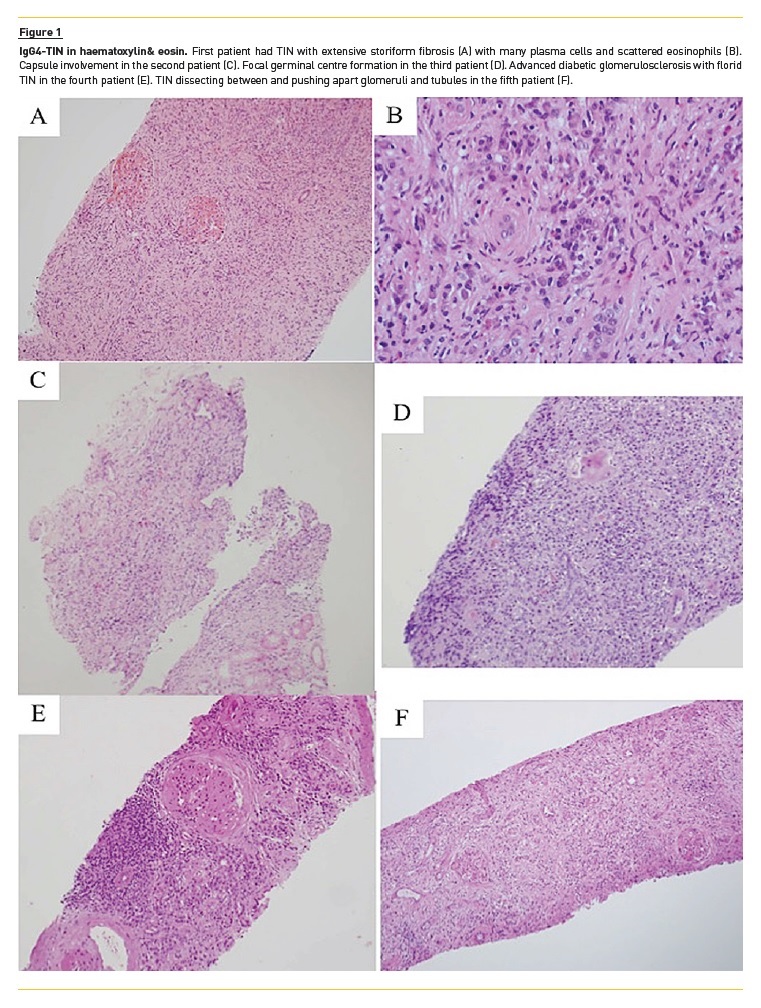
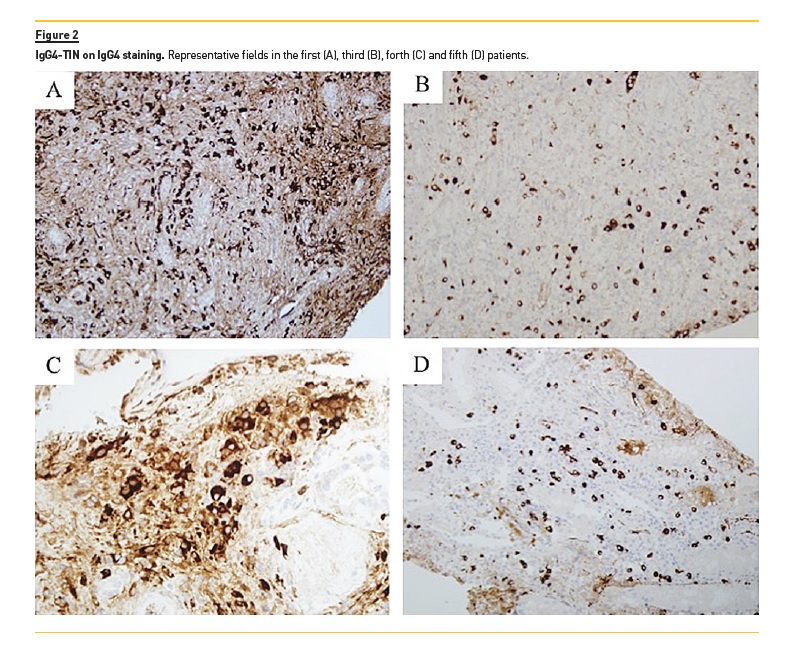
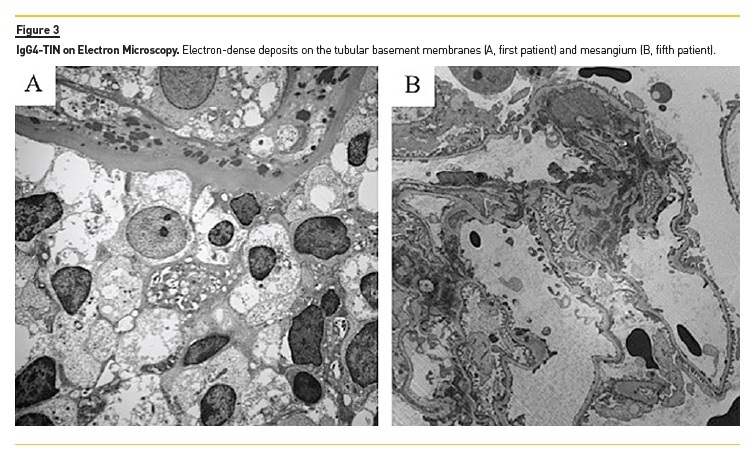
1st case (reported in 2013(1)): A 29‑year‑old man presented with lethargy, sinusitis and wheeze. He was found to have hypertension, active urinary sediment, a urine protein creatinine ratio (uPCR) of 2.4 g/g and renal dysfunction (serum creatinine‑SCr 3.5 mg/dL). He had antibodies to proteinase 3 by ELISA, elevated IgG4 (19.8 g/L) and normal C3 (98 mg/dL, normal range 70–170 mg/dL) and C4 (45 mg/dL, normal range 16–54 mg/dL). Renal ultrasound showed normal sized kidneys with multiple hyper‑echoic foci in the renal cortices. On MRI these areas had high T2 signal. Renal biopsy showed TIN with scattered eosinophils and extensive storiform fibrosis (Figure 1A and 1B). Many plasma cells were positive for IgG4 staining (Figure 2A). Electron‑dense deposits (EDD) on the tubular basement membranes (TBM) were present. Treatment with prednisolone 40 mg/day was started and renal function improved after 2 months (SCr 2.6 mg/dL). Follow‑up was then lost.
2nd case: A 60‑year‑old man, previously hypertensive and diabetic, presented with lymphadenopathy, sialadenitis, renal dysfunction (SCr 2.5 mg/dL) and proteinuria (uPCR 0.6 g/g). He had high IgG4 (2.3 g/L) and low C3 (56 mg/dL) and C4 (3 mg/dL) serum levels.
Lymph node and salivary gland biopsy were non‑specific. Renal biopsy showed TIN involving 15% of interstitium and extending into the capsule (Figure 1C).
The focus was not sampled on IgG4 staining. EDD on TBM were seen (Figure 3A). He was started on prednisolone 60mg/day with renal function and proteinuria improvement (SCr 1.5 mg/dL and uPCR 0.2 g/g).
3rd case: A 59‑year‑old diabetic man showed renal dysfunction (SCr 3.2 mg/dL) and proteinuria (uPCR 2.9 g/g), high IgG4 (1.7 g/L) and low C3 (54 mg/dL) and C4(5 mg/dL) levels. Renal biopsy demonstrated TIN with a focal germinal centre formation (Figure 1D) containing IgG4+ plasma cells (Figure 2B). TBM was thickened and had EDD. Treatment with prednisolone 60 mg/day resulted in renal function improvement (SCr 1.7 mg/dL) and complete proteinuria remission.
4th case: A 66‑year‑old man with a history of diabetes mellitus following what was thought to be an autoimmune pancreatitis, developed submandibular lymphadenopathy (IgG4+ plasma cells on biopsy), renal dysfunction (SCr 2.5 mg/dL) and heavy proteinuria (uPCR 8.8 g/g). He had high IgG4 level (16.7 g/L) and normal C3 and C4. PET‑CT showed low‑level activity in small nodes in neck and mediastinum. Renal biopsy demonstrated advanced diabetic glomerulosclerosis and a florid TIN (Figure 1E) with IgG4+ plasma cells (Figure 2C). Because there was marked tubulointerstitial scarring and limited and asymptomatic adenopathy, no specific treatment was offered. Six months later, SCr was 3.1 mg/dL and uPCR 9.8 g/g.
5th case: a 66‑year‑old man with renal impairment (SCr 2.6 mg/dL) and light proteinuria (uPCR 0.4g/g) was diagnosed with a reactive arthropathy and was treated with steroids. The renal function improved (SCr 2.0 mg/dL). Subsequent decline in renal function (while in steroids) was observed as well as low C3 (5 mg/dL) and C4 (7 mg/dL) and high IgG4 level (13.9 g/L). Renal biopsy showed a prominent TIN that dissected between and pushed apart glomeruli and tubules (Figure 1F). Many plasma cells were positive on IgG4 staining (Figure 2D).
There were mesangial EDD. Treatment with prednisolone 60 mg/day was recently restarted.
MINI‑REVIEW
IgG4‑RD is an emerging disease, challenging to diagnose and under‑recognized, particularly in patients with single‑organ system involvement. It is frequently diagnosed unexpectedly in pathological specimens or on imaging studies.
It was first described in 2001, when Hamano et al. reported high serum levels of IgG4 in patients with type 1 autoimmune pancreatitis (AIP). Two years later, the term IgG4‑related autoimmune disease was proposed after discovering that patients with AIP also had involvement of other organs. Several retrospective studies have revealed that IgG4‑RD accounts for many cases of idiopathic disease in diverse organs. For example, one review reported that 35% of patients with autoimmune hepatitis had features of IgG4‑RD (2).
Subsequently, the hepatology community has emphasized the identification of IgG4‑associated cholangitis distinguishing it from non IgG4‑related ones, since this has extremely important treatment implications(steroids versus surgery)3. Various other diseases previously described separately, such as Riedels thyroiditis, Küttnertumour (sialadenitis), and Mikuliczs syndrome (sialadenitis with dacryoadenitis), are currently considered part of the spectrum of IgG4‑RD. The proportion of IgG4‑TIN in all TIN remains unknown4.
The pathophysiology of IgG4‑RD is incompletely understood. Abnormal T helper type 2 (Th2)/T regulatory cells produce profibrotic cytokines leading to collagen deposition and fibrosis5,6.
Generally, IgG4‑RD affects middle‑aged to elderly patients with a clear male preponderance7‑9 but demography varies depending on the pattern of organ involvement (for example, sialadenitis is more frequent in young women). It can simulate other systemic conditions such as sarcoidosis, lymphoma, and Sjögrens syndrome and, when affecting a single‑organ (which happens in 40% of the cases9), it may be initially misdiagnosed as an autoimmune disorder such as nephritis, pancreatitis, or cholangitis.
By far the most common clinical manifestations are AIP, sialadenitis with dacryoadenitis, and retroperitoneal fibrosis8. Other involved sites include periorbital tissues, kidneys, lungs, thyroid, mediastinum, lymph nodes, meninges, aorta, prostate, skin and the pericardium.
Although multi‑organ disease is frequently identified at diagnosis, metachronous evolutions of the disease are well described, with the addition of one organ after another over periods of months to years10.
International Consensus Criteria for IgG4‑RD diagnosis were published in 2012 after a meeting of experts in Boston11. Diagnosing IgG4‑RD requires an adequate tissue biopsy demonstrating storiform fibrosis, mild to moderate tissue eosinophils, lymphoplasmacytic infiltrate, increased IgG4+ plasma cell levels – generally >50–100/ high power field (HPF) for most tissues – and an IgG4/IgG+ plasma cell ratio >40%. Obliterative venulitis is also a characteristic feature. Although a useful guide, the minimum IgG4+ plasma cell numbers recommended in the Boston histological criteria are based on small numbers of studies and many specialists advise that it should not be used definitively to exclude IgG4‑RD if the IgG4+ plasma cell numbers fall just short of the suggested minimum in a proper clinical context. Furthermore, measuring IgG4/IgG ratio has a reported sensitivity of 59%, a specificity of 90% and positive and negative likelihood ratios of 3.12 and 0.26, respectively12. Moreover, as with many other conditions apart from IgG4‑RD, histological changes are often patchy. Thus negative or inconclusive findings in these specimens can occur as a consequence of sampling (as occurred with our second patient). On the other hand, these diagnostic cutoff points vary depending on which organs are affected and diagnosis may be more complex in advanced cases, where extensive fibrosis with only a few plasma cells are the main features.High serum IgG4 (greater than 1.3 5g/L) is important and there is a correlation between serum IgG4 and the number of involved organs10. However, it is not sensitive since 30–45% of patients with confirmed IgG4‑RD have normal serum IgG4 levels (mainly in patients with single organ involvement)8,13.
Serum IgG4 levels are also not specific since moderately elevated levels are seen in numerous inflammatory, autoimmune and neoplastic conditions8,13. Nevertheless, extremely high serum IgG4 levels (>10 g/L) are rarely seen outside IgG4‑RD8,13. Other important serum finding is IgE level, which is elevated in approximately 60% of the patients10. Antinuclear antibodies and rheumatoid factor are positive in 20–30% of the patients14.
RENAL INVOLVEMENT
It is estimated that 12% of patients with IgG4‑RD will have renal parenchyma involvement(8). The Japanese Nephrology Society biopsy registry reported that the number of histological diagnoses for IgG4‑RKD was less than one‑tenth that of rapid progressive glomerulonephritis and less than one‑half that of renal amyloidosis15.
IgG4‑RD frequently involves structures adjacent to the kidney, making obstructive uropathy secondary to retroperitoneal fibrosis the most common renal involvement of IgG‑RD.
In fact, a large review of 125 IgG4‑RD patients reported that retroperitoneal fibrosis accounted for 18% (n=23) of renal failure cases8. Rarely, the renal pelvis can be affected in the form of IgG4 pyelitis.
As previous highlighted, 12% of IgG4‑RD patients have renal intrinsic involvement (IgG4‑KD).
This occurs mainly as tubulo‑interstitial nephritis (IgG4‑TIN) but membranous nephropathy, anti‑PLA2R negative (with or without TIN), is responsible for 10% of the cases16‑18.
Moreover, diabetes mellitus secondary to pancreas involvement (which may be the cause of diabetes in our patients) can lead to diabetic nephropathy (with or without TIN), as probably happened with our fourth patient.
Patients with kidney involvement (isolated or with other organ disease) are most commonly middle aged to elderly with a male preponderance7,19. In 83–98% of the cases, IgG4‑TIN occurs with extra‑renal manifestations19–21.
Renal disease has a wide spectrum of presentations, ranging from normal to severely compromised renal function in the form of acute or chronic renal failure. Distinctively from other TIN, IgG4‑TIN has no urinary white blood cells or white blood cells casts19 and usually it does not present with elevated systemic acute phase reactants (such as C‑reactive protein), since tissue inflammation is not accompanied by systemic inflammation19.
The great majority of patients with IgG4‑TIN have elevated serum IgG and/or IgG4 levels, with reported rates of 70–88%19,20.
Strikingly, three of our five patients (60%) had hypocomplementemia (both C3 and C4). This is consistent with literature reports, which describe that more than half of patients with renal involvement present hypocomplementemia (usually both low C3 and C4), as opposed to those with IgG4‑RD without renal disease (56% vs. 20% according to Mayo Clinic assessment20).
This fact suggests that complement plays a role in the pathogenesis of this disorder and more studies are needed to elucidate the underlying pathways.
Regarding imaging exams, usually enlarged kidneys or mass lesions can usually be seen. However, atrophic kidneys are the main features in the later stages of the disease. Typical features on contrast CT scan are low‑enhancing, small, bilateral, nodular or wedge‑shaped lesions, diffusely or in the peripheral cortex22. Importantly, these lesions are not visible on non‑contrast CT scan22. On MRI they are isointense on T1‑weighted and low‑enhancing in T2‑weighted with diffusion restriction, and a progressive enhancement pattern22,23. As stated before, less frequently the lesions may be extra‑parenchymal (for instance in the renal sinus), mimicking masses or cysts22. For diagnosing IgG4‑KD lesions, sensitivity of MRI with diffusion‑weighted imaging (DWI) is estimated to be 100%, which is much higher than the 77% found for T2‑weighted MRI23. 18‑F‑fluorodeoxyglucose positron emission tomography/computed tomography can show diffuse moderate uptake and enlargement of involved organs and it may become a useful tool for assessing organ involvement, monitoring therapeutic response, and guiding interventional treatment of IgG4‑RD24.
Despite the importance of previous clinical and imaging features, the definitive IgG4‑RD diagnosis is often made by histology.
In IgG4‑TIN, the common diagnostic features are (A) diffuse or multifocal TIN, with a variable ratio of fibrosis (typically in a storiform pattern) to inflammation, (B) moderate to marked interstitial increase in IgG4+ plasma cells and (C) increased mononuclear cells with eosinophils in most cases11,16,20. Occasionally focal and mild mononuclear cell tubulitis can be seen, as opposed to plasma cell tubulitis which is rarely present. Glomeruli are normal or have mild mesangial matrix expansion.
In IgG4‑membranous nephropathy granular subepithelial deposits may be seen by immunofluorescence, electron microscopy, or light microscopy including immunoperoxidase staining for IgG418. TBM immune complex deposits, made up of polyclonal IgG and C3, are often noted.
Considering that solely looking for plasma cell infiltration would lead to over‑diagnosis and simply looking at biopsies with moderate to severe interstitial inflammation would lead to under‑diagnosis, Mayo Clinic conducted a 1‑year review of 414 renal biopsies with moderate/severe inflammation20. The authors found that 20% of the cases had increased plasma cells but of those only 2% were IgG4‑TIN.
Therefore, the Mayo Clinic proposed an IgG4‑TIN diagnostic criteria scaffold (Table I), which is complementary to International Consensus Criteria for general IgG4‑RD.
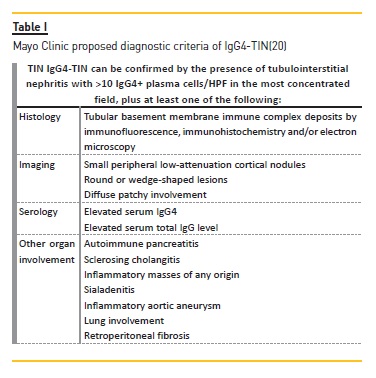
This group stated that when IgG4‑TIN is suspected (following clinical, serology or imaging features), a lymphoplasmacytic infiltrate should be searched for in renal biopsy and if increased plasma cells are present these should be stained for IgG and IgG4. These stains can also be performed on previous archived samples from kidney and other organs, particularly if they were classified as non‑diagnostic biopsies. The authors also demonstrated that after excluding pauci‑immune necrotizing and crescentic glomerulonephritis, IgG4 immunohistochemistry had a sensitivity of 100% and a specificity of 92% for IgG4‑TIN when using a cut‑of of >10 plasma cells/HPF.
Other diagnoses to be considered and ruled out are medication‑induced interstitial nephritis, Systemic Lupus Erythematous (SLE), Sjögrens syndrome, other autoimmune TIN and chronic pyelonephritis.
It is important to keep in mind that the histologic pattern of TIN is related to the stage of the disease: (A) acute TIN with minimal fibrosis, (B) a more cellular inflammatory pattern with storiform fibrosis, or (C) paucicellular fibrosis20.
The 2015 International Consensus Statement recommended to treat patients with active IgG4‑RD urgently and states that many patients with asymptomatic disease may require treatment to prevent or minimize future organ damage25. On the other hand, patients with extensive fibrosis (as seen in our fourth patient) may not benefit from therapy.
The first‑line treatment for IgG4‑RD, including isolated IgG4‑TIN, is glucocorticoids. The typical regimen is prednisone 0.6–1 mg/day over 4 weeks followed by a dose reduction to 5 mg/day in six months. A prospective Japanese single‑arm study of 57 patients reported an overall response rate of 82% with a relapse rate of 12% with this regimen26. New or worsening diabetes was the foremost toxicity, occurring in 30% of patients. Other studies show similar renal recovery rates21,27,28.
Since 2010, B cell depleter rituximab has also been used successfully in IgG4‑RD.
An American prospective, single‑arm trial using 1g intravenous rituximab 2 weeks apart, demonstrated a clinical response in 29 of 30 patients, of whom 7 (24%) relapsed29. Moreover, rituximab has been effective in most cases refractory to steroids.
Regarding typical steroid‑sparing agents (azathioprine, mycophenolate mofetil, methotrexate, tacrolimus or cyclophosphamide), they have been used mainly in AIP but with limited success. However, there are case reports of successful IgG4‑TIN treatment with low dose of prednisolone and azathioprine or mizoribine30.
The majority of patients with IgG4‑TIN show improvement of renal function and radiologic lesions as soon as treatment is started. Then a plateau stage is observed after weeks to months or a slight decline may occur.
Notably, even patients with tubulointerstitial fibrosis and estimated glomerular filtration rate less than 60ml/min can partially recover renal function as non‑fibrotic areas recover28.
However, IgG4‑RD is typically a chronic disease with frequent relapse (20–40%) and regression of the disease in one organ may be followed by development of the disease in another. Therefore, frequently, maintenance steroids (prednisolone 2.5–5 mg/day) or re‑treatment with rituximab are needed25.
Besides regular renal function tests, increased serum IgG4, low C3 andC4 and proteinuria are helpful markers to predict a relapse, particularly if present initially21, and they should be monitored closely in order to plan for re‑treatment at the earliest sign of relapse. Quantitative flow cytometry of peripheral blood plasmablasts (CD19low CD38+ CD20‑CD27+) has been proposed as a more sensitive biomarker for diagnosis, assessing response to treatment, and determining the appropriate time for re‑treatment31. However, this exam is not available in most centers.
The proportion of patients with end‑stage renal disease secondary to IgG4‑TIN remains unknown. Data on renal transplant is scant.
CONCLUSIONS
In brief, the relevance of recognizing IgG4‑RD resides in its recent discovery by the medical community and in the implications it may have in causing organ failure when not diagnosed early enough. Thus, IgG4‑RKD should be considered in patients presenting with abnormal urinalysis, renal dysfunction, renal lesions on imaging and elevated IgG and/or IgE or hypocomplementemia.
Diagnosis of IgG4‑TIN requires a combination of histologic features (plasma cell‑enriched TIN with >10 IgG4+ plasma cells/HPF, with/without TBM immune complex deposits in many cases) and at least one of the following: (A) characteristic radiologic findings (small peripheral low‑signal cortical nodules, round or wedge‑shaped lesions, or diffuse patchy involvement), (B) elevated serum IgG4 level or (C) characteristic findings of IgG4‑RD in other organs. Other conditions that also present with IgG4+ plasma cells in the renal parenchyma (SLE, vasculitis and lymphoma) and other causes of TIN (drugs and infections) should be excluded. Since IgG4‑TIN has a non‑specific histologic appearance, the pathologist should be specifically informed of a clinical suspicion for IgG4‑RD to conduct proper immunohistochemical evaluation. IgG4‑RD can be treated with steroids and/or rituximab, and the highly variable degrees of recovery depend on the extent of fibrosis at the time of diagnosis. Relapses are frequent and patients demand close long‑term follow‑up.
References
1. Sayed R, Cook T, Palmer A. Recognizing isolated IgG4‑related nephropathy. Clin Kidney J. 2013;6(4):433‑5. [ Links ]
2. Chung H, Watanabe T, Kudo M, Maenishi O, Wakatsuki Y, Chiba T. Identification and characterization of IgG4‑associated autoimmune hepatitis. Liver Int. 2010;30(2):222‑31. [ Links ]
3. Joshi D, Webster GJ. Immunoglobulin G4‑related sclerosing cholangitis. Hepatology. 2015;61(4):1432‑4. [ Links ]
4. Mann S, Seidman MA, Barbour SJ, Levin A, Carruthers M, Chen LY. Recognizing IgG4‑related tubulointerstitial nephritis. Can J Kidney Health Dis. 2016;3:34. [ Links ]
5. Zaidan M, Cervera‑Pierot P, de Seigneux S, Dahan K, Fabiani B, Callard P, et al. Evidence of follicular T‑cell implication in a case of IgG4‑related systemic disease with interstitial nephritis. Nephrol Dial Transplant. 2011;26(6):2047‑50. [ Links ]
6. Umehara H, Nakajima A, Nakamura T, Kawanami T, Tanaka M, Dong L, et al. IgG4‑related disease and its pathogenesis‑cross‑talk between innate and acquired immunity. Int Immunol. 2014;26(11):585‑95. [ Links ]
7. Stone JH, Zen Y, Deshpande V. IgG4‑related disease. N Engl J Med. 2012;366(6):539‑51. [ Links ]
8. Wallace ZS, Deshpande V, Mattoo H, Mahajan VS, Kulikova M, Pillai S, et al. IgG4‑Related Disease: Clinical and Laboratory Features in One Hundred Twenty‑Five Patients. Arthritis Rheumatol. 2015;67(9):2466‑75. [ Links ]
9. Brito‑Zeron P, Ramos‑Casals M, Bosch X, Stone JH. The clinical spectrum of IgG4‑related disease. Autoimmun Rev. 2014;13(12):1203‑10. [ Links ]
10. Stone JH, Brito‑Zeron P, Bosch X, Ramos‑Casals M. Diagnostic approach to the complexity of IgG4‑related disease. Mayo Clin Proc. 2015;90(7):927‑39. [ Links ]
11. Deshpande V, Zen Y, Chan JK, Yi EE, Sato Y, Yoshino T, et al. Consensus statement on the pathology of IgG4‑related disease. Mod Pathol. 2012;25(9):1181‑92. [ Links ]
12. Deng C, Li W, Chen S, Zhang W, Li J, Hu C, et al. Histopathological diagnostic value of the IgG4+/IgG+ ratio of plasmacytic infiltration for IgG4‑related diseases: a PRISMA‑compliant systematic review and meta‑analysis. Medicine (Baltimore). 2015;94(9):e579. [ Links ]
13. Carruthers MN, Khosroshahi A, Augustin T, Deshpande V, Stone JH. The diagnostic utility of serum IgG4 concentrations in IgG4‑related disease. Ann Rheum Dis. 2015;74(1):14‑8. [ Links ]
14. Umehara H, Okazaki K, Masaki Y, Kawano M, Yamamoto M, Saeki T, et al. A novel clinical entity, IgG4‑related disease (IgG4RD): general concept and details. Mod Rheumatol. 2012;22(1):1‑14. [ Links ]
15. Nakashima H, Kawano M, Saeki T, Ubara Y, Hisano S, Nagata M, et al. Estimation of the number of histological diagnosis for IgG4‑related kidney disease referred to the data obtained from the Japan Renal Biopsy Registry (J‑RBR) questionnaire and cases reported in the Japanese Society of Nephrology Meetings. Clin Exp Nephrol. 2017;21(1):97‑103. [ Links ]
16. Saeki T, Kawano M. IgG4‑related kidney disease. Kidney Int. 2014;85(2):251‑7. [ Links ]
17. Cortazar FB, Stone JH. IgG4‑related disease and the kidney. Nat Rev Nephrol. 2015;11(10):599‑609. [ Links ]
18. Alexander MP, Larsen CP, Gibson IW, Nasr SH, Sethi S, Fidler ME, et al. Membranous glomerulonephritis is a manifestation of IgG4‑related disease. Kidney Int. 2013;83(3):455‑62. [ Links ]
19. Kawano M, Saeki T. IgG4‑related kidney disease: an update. Curr Opin Nephrol Hypertens. 2015;24(2):193‑201. [ Links ]
20. Raissian Y, Nasr SH, Larsen CP, Colvin RB, Smyrk TC, Takahashi N, et al. Diagnosis of IgG4‑related tubulointerstitial nephritis. J Am Soc Nephrol. 2011;22(7):1343‑52. [ Links ]
21. Saeki T, Kawano M, Mizushima I, Yamamoto M, Wada Y, Nakashima H, et al. The clinical course of patients with IgG4‑related kidney disease. Kidney Int. 2013;84(4):826‑33. [ Links ]
22. Takahashi N, Kawashima A, Fletcher JG, Chari ST. Renal involvement in patients with autoimmune pancreatitis: CT and MR imaging findings. Radiology. 2007;242(3):791‑801. [ Links ]
23. Kim B, Kim JH, Byun JH, Kim HJ, Lee SS, Kim SY, et al. IgG4‑related kidney disease: MRI findings with emphasis on the usefulness of diffusion‑weighted imaging. Eur J Radiol. 2014;83(7):1057‑62. [ Links ]
24. Zhang J, Chen H, Ma Y, Xiao Y, Niu N, Lin W, et al. Characterizing IgG4‑related Disease with (1)(8)F‑FDG PET/CT: a prospective cohort study. Eur J Nucl Med Mol Imaging. 2014;41(8):1624‑34. [ Links ]
25. Khosroshahi A, Wallace ZS, Crowe JL, Akamizu T, Azumi A, Carruthers MN, et al. International consensus guidance statement on the management and treatment of IgG4‑related disease. Arthritis Rheumatol. 2015;67(7):1688‑99. [ Links ]
26. Masaki Y, Shimizu H, Sato Nakamura T, Nakamura T, Nakajima A, Iwao Kawanami H, et al. IgG4‑related disease: diagnostic methods and therapeutic strategies in Japan. J Clin Exp Hematop. 2014;54(2):95‑101. [ Links ]
27. Saeki T, Kawano M, Mizushima I, Yamamoto M, Wada Y, Ubara Y, et al. Recovery of renal function after glucocorticoid therapy for IgG4‑related kidney disease with renal dysfunction. Clin Exp Nephrol. 2016;20(1):87‑93. [ Links ]
28. Mizushima I, Yamada K, Fujii H, Inoue D, Umehara H, Yamagishi M, et al. Clinical and histological changes associated with corticosteroid therapy in IgG4‑related tubulointerstitial nephritis. Mod Rheumatol. 2012;22(6):859‑70. [ Links ]
29. Carruthers MN, Topazian MD, Khosroshahi A, Witzig TE, Wallace ZS, Hart PA, et al. Rituximab for IgG4‑related disease: a prospective, open‑label trial. Ann Rheum Dis. 2015;74(6):1171‑7. [ Links ]
30. Miyata KN, Kihira H, Haneda M, Nishio Y. IgG4‑related tubulointerstitial nephritis associated with membranous nephropathy in two patients: remission after administering a combination of steroid and mizoribine. Case Rep Nephrol. 2014;2014:678538. [ Links ]
31. Wallace ZS, Mattoo H, Carruthers M, Mahajan VS, Della Torre E, Lee H, et al. Plasmablasts as a biomarker for IgG4‑related disease, independent of serum IgG4 concentrations. Ann Rheum Dis. 2015;74(1):190‑5. [ Links ]
Joana Rocha, MD
Department of Nephrology, Centro Hospitalar de Tras‑os‑Montes e Alto Douro, Av. Noruega, Lordelo, 5000‑508 Vila Real, Portugal.
Tel: +351 914118060.
Email: joanafvrocha@gmail.com
Disclosure of potential conflicts of interest: none declared.
Authors Contributions: Joana Rocha collected the clinical and histopathological data, conducted the literature review and drafted the article. Amy Wang and Nikki Wong participated in clinical data collection. Joseph Westaby participated in histopathological review of the cases. Candice Roufosse and Terence Cook performed critical revision of the article. All the authors gave final approval of this version to be published.
Received for publication: Aug 30, 2017
Accepted in revised form: Oct 30, 2017


{kind=link}
{kind=link}
{kind=link}