










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
versão impressa ISSN 0872-0169
Port J Nephrol Hypert vol.32 no.1 Lisboa mar. 2018
REVIEW ARTICLE
Off-target effects and adverse outcomes of fibroblast growth factor 23 in chronic kidney disease
Giovana S. Di Marco, Marcus Brand
Department of Internal Medicine D, University Hospital Muenster, Muenster, Germany
ABSTRACT
Chronic kidney disease (CKD) patients have a high risk of death, especially cardiovascular death. High fibroblast growth factor 23 (FGF23) levels are independently associated with higher risk of death and cardiovascular events in these patients, as well as with a greater risk of severe inflammation. Chronic inflammation contributes to the progression of renal disease, the pathogenesis of cardiovascular disease, and mortality. Moreover, clinically, the CKD-associated defect in the immune system also has a relevant impact on morbidity and mortality. Recent findings showing that excess FGF23 may affect non-traditional, off-target organs independently of αKlotho give support to the potential clinical relevance of the adverse effects of FGF23 in CKD, and even in non-CKD patients. In this review, we provide an update on FGF23 and briefly discuss its future in clinical practice.
Keywords: Chronic kidney disease (CKD), fibroblast growth factor 23 (FGF23), heart hypertrophy, inflammation.
INTRODUCTION
Chronic kidney disease (CKD) is associated with an increased risk of death, cardiovascular events, and hospitalization.1,2 These adverse outcomes, however, cannot be fully explained by the presence of known, traditional risk factors.2 Indeed, non-traditional risk factors appear to be particularly relevant to patients with CKD. These CKD-associated risks include oxidative stress, endothelial dysfunction, decreased hemoglobin levels, and progression of CKD itself.3,4 Other non-traditional risk factors of great interest are the presence of inflammation, which is frequently activated in CKD patients, and abnormalities in bone and mineral metabolism. The regulation of calcium and phosphate was only partially understood until the discovery of fibroblast growth factor 23 (FGF23) around the 2000s.5 Meanwhile, however, much attention has been paid to FGF23s multiple actions, which go beyond mineral metabolism and place FGF23 as a potential clinical marker or mediator associated with adverse outcomes in CKD as well as non-CKD patients.
UNDERSTANDING FGF23S ACTIONS AND REGULATIONS
FGF23 is one of 22 members of the FGF family of proteins that bind and activate alternatively spliced forms of four tyrosine kinase FGF receptors (FGFR1-4).6 Most FGFs are paracrine/autocrine factors or function as intracellular mediators, while FGF23 is an endocrine hormone. In fact, intracellular FGFs exert their functions in a receptor-independent manner while canonical FGFs have a heparin-binding site that is necessary for stable binding to the receptors and local signaling. Endocrine FGFs function as circulating factors owing to their reduced heparin/heparan sulfate-binding affinity, which prevents them from being captured in the extracellular matrix.
On the other hand, this reduced affinity also prevents direct interactions between the FGFs and their receptors.7 Endocrine FGFs overcome this handicap by using Klotho proteins instead of heparin-sulfate to enhance receptor binding. FGF23 uses αKlotho as a cofactor,8 a molecule that, other than serving as a co-receptor for FGF23, also circulates as an endocrine substance and exerts a multitude of important effects.5 Considering the ubiquity of FGFRs and the restricted expression of αKlotho, it seems that the presence of αKlotho governs whether a cell is an FGF23 target.9
The primary physiological function of FGF23 is to regulate phosphate homeostasis in the kidneys.10
FGF23 suppresses phosphate reabsorption by binding FGFR-Klotho complexes, which activates the mitogen-activated protein kinase (MAPK) signaling cascade leading to downregulation of sodium-dependent phosphate co-transport (NaPi-2a) in the proximal tubule.11,12 This effect is predominantly mediated by the activation of FGFR1 signaling mechanisms, but also to a lesser extent by activation of FGFR4.13,14 In addition, FGF23 suppresses renal synthesis of the active form of vitamin D, 1,25-dihydroxyvitamin D [1,25(OH)2D] by inhibiting the expression of the activating enzyme 25-dydroxyvitamin D-1α-hydroxylase and, simultaneously, stimulating the catabolic enzyme D-24-hydroxylase.15 FGF23 also reduces the production of parathyroid hormone (PTH) by activating a Klotho-independent calcineurin-mediated signaling pathway in the parathyroid glands.16
In individuals with normal kidney function, increased circulating FGF23 levels lead to renal phosphate wasting and, subsequently, impaired bone mineralization.
In the early stages of reduced renal function, a compensatory increase in FGF23 levels helps to maintain phosphate balance,17 but at the cost of calcitriol deficiency, secondary hyperparathyroidism, and perhaps Klotho deficiency.18 In advanced stages of CKD, these compensatory mechanisms become overwhelmed and blood FGF23 concentrations increase drastically over phosphate and PTH levels, eventually peaking during dialysis, thus resulting in a group of abnormalities referred to as CKD-mineral bone disease.19
FGF23 is predominately secreted by bones (osteocytes and osteoblasts) in response to several physiologic stimuli, including 1,25(OH)2D, PTH, and phosphate load.20 However, since an increase in FGF23 production precedes the increase of PTH, and both FGF23 and PTH increase before hyperphosphatemia first appears,17 the primary stimulus for enhanced FGF23 production in early CKD is still unknown. It is noteworthy that several other factors have been described to directly or indirectly stimulate FGF23 production, including aldosterone, serum calcium, pro-inflammatory cytokines, and parameters of iron metabolism.20 Since FGF23 is a small 32-KDa protein, a reduced glomerular filtration rate may concomitantly reduce renal elimination of FGF23 and contribute to increased blood levels of FGF23.13,21 In addition, under pathological circumstances, FGF23 may also be produced by extra-skeletal sources, including the kidneys, cells of the innate immune system (e.g. macrophages), cardiomyocytes, bone marrow, and erythroid cells.21-25
It is not clear, however, to what extent extra-skeletal-derived FGF23 would add to high circulating FGF23 levels. However, increased local production may also be able to stimulate receptor-mediated signaling,24 thereby acting in a paracrine manner.
Some of the FGF23 synthesized by osteocytes is processed by furin, a proteolytic enzyme, yielding Nand C-terminal protein fragments.26,27 This intracellular cleavage abrogates the phosphaturic actions of FGF23 and is considered the most important mechanism of FGF23 regulation.28 The two forms, intact or full-length FGF23 (iFGF23; ∼32 kDa) and the C-terminal fragment (cFGF23, ∼14 kDa) can be detected in human plasma and exert different biological activities.29,30
Commercially available assays for cFGF23 detect both cFGF23 and iFGF23, while the iFGF23 assay detects only the intact hormone. C-terminal FGF23 is less abundant in CKD patients than in control subjects and is found at very low levels in dialysis patients.29,31
Excess FGF23 may be caused by a combination of increased FGF23 production and defective cleavage.
Thus, with the progression of renal disease, it is conceivable that FGF23 production simply overcomes cleavage.29 C-terminal FGF23 competes with iFGF23 for receptor binding, thereby antagonizing its phosphaturic activity.14 Indeed, some studies have shown a significant association of iFGF23 – but not cFGF23 – with parameters such as phosphate32 and insulin growth factor,33 whereas others have demonstrated significant association of cFGF23 – but not iFGF23 – with, for example, iron34 and phosphate.33 Despite these discrepancies, higher iFGF23 and cFGF23 levels have both been consistently and independently associated with mortality risk and decline in renal function in patients with CKD.35-37
PATHOLOGICAL FGF23 LEVELS AND OFF-TARGET EFFECTS
Pathologically elevated levels of FGF23 may exert αKlotho-independent effects on non-traditional, off-target organs, such as the heart, liver, and cells of the immune system. These maladaptive effects may thereby contribute to cardiovascular disease, kidney disease progression, and mortality, not only in CKD patients, but also in non-CKD populations (Figure 1).
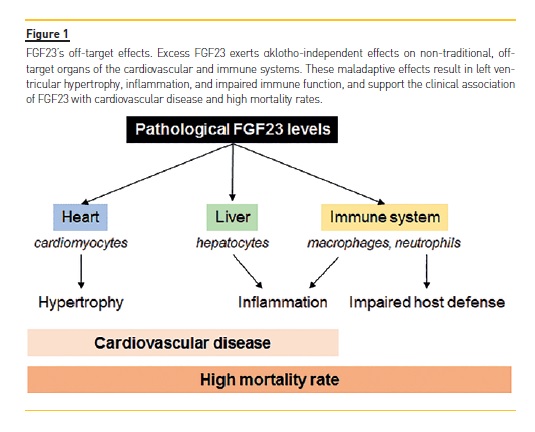
In a groundbreaking study, Faul et al. showed that FGF23 can cause hypertrophic growth of individual cardiac myocytes in vitro and induce left ventricular hypertrophy (LVH) in mice via FGFR-dependent and αKlotho-independent activation of the calcineurin-nuclear factor of activated T cells (NFAT) signaling cascade, which is known to mediate pathological cardiac hypertrophy in response to other pathogenic factors.38,39 Afterwards, Grabner et al. identified FGFR4 as the cardiac receptor for FGF23.40 In proof-of-principle studies, blocking FGFR with a pan-FGFR inhibitor attenuated LVH in a rat model of CKD, despite sustained renal impairment and severe hypertension.38,41 These findings support FGFR activation as a potentially modifiable, blood pressure- and volume-independent molecular mechanism of LVH in CKD. However, although these studies demonstrated cardioprotective effects of FGFR blockade in the absence of any changes in serum phosphate or tissue mineralization, 41 the toxicity of pan-FGFR inhibitors precludes this approach in humans.15,42,43 Similarly, monoclonal antibodies that neutralize FGF23 seem to be of limited utility in CKD because complete abrogation of FGF23s effects can cause severe hyperphosphatemia, inducing diffuse vascular calcification and increased mortality.43 The independent effects of FGF23 on the kidneys via FGFR1 and on the heart via FGFR4 hold promise for future clinical therapeutic and drug development, given that selective inhibition of FGFR444 would block the toxic effects of FGF23 in the heart without interfering with the essential effects of FGF23 in the regulation of phosphate homeostasis.
Accordingly, Grabner et al. have shown that a specific FGFR4-blocking antibody is able not only to prevent the development of LVH but also to reverse established LVH in rats with CKD.40,45
In fact, FGF23 exhibits off-target effects via FGFR4 not only on the myocardium, but also on the liver. Fauls group demonstrated through a series of in vitro and in vivo experiments that FGF23 binds to hepatic FGFR4 and, in an αKlotho-independent and NFAT-dependent manner, directly induces hepatic expression and secretion of inflammatory cytokines such as interleukin 6 and C-reactive protein (CRP).46 As a proof-of-concept, pharmacologic FGFR4 blockade or calcineurin inhibition reduced hepatic and systemic CRP levels in CKD rats.46
Immune cells are also targets of FGF23. In macrophages, FGF23 exerts its functions via FGFR1. FGF23 secreted by pro-inflammatory, classically activated M1 macrophages acts in a paracrine manner to increase TNFα production in uncommitted M0 macrophages.
Moreover, FGF23 increases the number of macrophages and inhibits the anti-inflammatory functions of alternatively activated M2 macrophages. As cardiovascular disease and renal failure progression in CKD are associated with macrophage tissue infiltration, it is possible that the production of FGF23 by macrophages contributes to cardiac and renal fibrosis.47 Furthermore, the fact that FGF23 regulates important genes involved in inflammation and renal fibrosis (e.g. TGF-β and TNF-α) in murine models of CKD provides a possible mechanistic link between elevated FGF23 and pathways responsible for renal failure progression and cardiovascular diseases.48
While on the one hand FGF23 may induce tissue inflammation, on the other hand, it impairs leukocyte/neutrophil activation and recruitment into the inflamed tissue, thereby impairing bacterial clearance and host defense. Mechanistically, FGF23 inhibits integrin activation in neutrophils by signaling through FGFR2 independently of αKlotho. As a proof-of-concept, FGF23 blockade with a neutralizing antibody in a murine model of CKD restored recruitment and host defense in these animals. In ex vivo analysis, distinct impairments in several steps of the leukocyte recruitment cascade were also observed in samples from patients with CKD. Incubation of healthy control leukocytes with FGF23 completely abrogated neutrophil slow rolling, while treatment of patients leukocytes with an FGFR inhibitor was able to rescue chemokine-induced arrest in these cells.49 Clinically, the CKD-associated defect in the immune system has a relevant impact on morbidity and mortality.50
Interestingly, the relationship between FGF23 and inflammation seems to be bidirectional. FGF23 induces inflammation, which, in turn, directly or indirectly stimulates FGF23 production.46,51,52 The direct effects are mediated by pro-inflammatory cytokines while the indirect effects are associated with alterations of well-known regulators of FGF23 production, including calcium, phosphate, and vitamin D metabolism. However, the major indirect effect of inflammation on FGF23 production seems to be associated with the regulation of iron metabolism. It is well-known that iron levels are reduced in the systemic circulation during inflammatory processes and hypoferremia leads to increased FGF23 production. 20,52 Iron deficiency stimulates fgf23 transcription in osteocytes but does not cause hypophosphatemia in wild-type mice because of increased intracellular degradation of FGF23, which results in elevation of circulating cFGF23 levels but normal iFGF23 levels.53
Such a causal nexus between inflammation and FGF23 production may contribute to the 100-to 1000-fold increase in FGF23 levels frequently found in dialysis patients, as well as to adverse outcomes in CKD patients.
Chronic inflammation contributes to the progression of renal disease, the pathogenesis of cardiovascular disease, and mortality.54 Consistently, higher FGF23 levels are independently associated with a greater risk of manifesting severe inflammation in patients with CKD,46,55,56 as well as with higher risks of death and cardiovascular events in multiple CKD and non-CKD populations.35,57-59
FGF23 IN CLINICAL PRACTICE
As a potential underlying mechanism for the increased risk of cardiovascular disease and death, epidemiological studies have consistently identified an independent association between elevated FGF23 levels and greater left ventricular mass as well as higher prevalence and incidence of LVH in a variety of populations (Figure 2).60,61 In support of the potential clinical relevance of adverse effects of FGF23 on the heart, high FGF23 levels are also independently predictive of higher risk of congestive heart failure events in both CKD and non-CKD patients.62,63 Given the strong relationships between LVH, congestive heart failure, and death,35,57,58,60-63 it is intriguing to speculate that FGF23 may contribute causally to adverse cardiovascular outcomes in CKD. FGF23 is elevated in the vast majority of CKD patients.
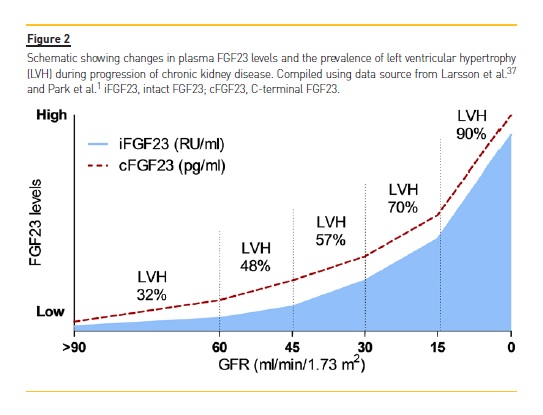
A prospective case-cohort study as part of the Chronic Renal Insufficiency Cohort (CRIC) Study that applied serial FGF23 measurements in patients with moderate to severe CKD showed that FGF23 levels were elevated but remained stable over time in the majority of the studied patients, while smaller subpopulations presented with slowly or rapidly rising FGF23 levels and a dramatically higher risk of death (4.49- and 15.23-fold higher, respectively).64
These observations and the mechanistic studies described above raise the question of whether an intervention to reduce FGF23 levels or inhibit its activity would improve outcomes in patients with CKD. The findings from preclinical studies with various therapies that target FGF23, including FGFR inhibitors, calcitriol/vitamin D,65 and anti-FGF23 neutralizing antibodies are supportive,49 but the effects of FGF2338,41,45 antagonism in CKD are still controversial.43
In humans, cinacalcet therapy lowers circulating FGF23 levels.66-68 In a secondary analysis of the Evaluation of Cinacalcet HCl Therapy to Lower Cardiovascular Events (EVOLVE) trial, Moe et al. showed that about 64% of the patients randomized to cinacalcet presented at least a 30% decrease in FGF23 levels; moreover, this decrease was associated with lower rates of cardiovascular events, including sudden cardiac death, and death. Whether or not these associations are a direct or indirect effect of cinacalcet remains to be clarified. However, these effects seem to be independent of demographic factors, comorbidities, and the cumulative dose of vitamin D sterols. In addition, even though a more pronounced reduction in FGF23 levels was observed in patients with blood levels of calcium, phosphate, and PTH that fell within the recommended range for clinical practice, cinacalcet lowers FGF23 even after accounting for changes in these parameters.67,68 In the placebo group, only 28% of the patients achieved such a reduction in FGF23 levels, and this reduction was not associated with a reduction in cardiovascular end-points.
While on the one hand cinacalcet is able to reduce FGF23, on the other hand, calcimimetics are not routinely available for predialysis patients with CKD. Dietary restriction of phosphate and phosphate binders appears to be a reasonable option to target FGF23.
Although controversial, reports on the FGF23-lowering effect of the various types of phosphate-binding therapies favor the use of sevelamer, while calcium-based phosphate binders may not reduce, and may even increase, FGF23 levels.69-71
Another possibility is iron therapy. In CKD patients with iron deficiency, switching the phosphate binder sevelamer to an oral iron supplement reduced circulating FGF23 levels independently of phosphate and vitamin D (other phosphate binders and cinacalcet were not changed).72 In addition, Wolf et al. have shown that intravenous iron in women with iron deficiency anemia lowers FGF23, whereas carbohydrate moieties in certain iron preparations transiently increase FGF23 by inhibiting its degradation.53 Here, it is worth noting that, concerning iron metabolism/supplementation, there are important differences regarding iFGF23 and cFGF23 levels and associations.34,53
Along these lines, even though FGF23 holds potential as a therapeutic target, data on the extent of its effects, the potency of the compounds to achieve significant reductions in FGF23 levels in different degrees of renal dysfunction, and the presence of a dose-effect relationship are still missing. Mechanistic aspects of how FGF23 levels increase in different stages of CKD and how these possible therapies lower FGF23 should also be explored in more depth. Furthermore, it is possible that important biological signals and clinical associations are being missed or underestimated owing to differences in iFGF23 and cFGF23 measurements, which strongly indicates the need for the establishment and standardization of reliable assays.
All this knowledge is, however, mandatory to determine whether reductions in FGF23 levels are directly responsible for improved clinical outcomes in CKD patients or if the observed benefits simply rely on better management of modifiers of cardiovascular and/or kidney disease in these patients.
Additional specific strategies for controlling FGF23 should be considered and adequately designed clinical trials are still needed to validate FGF23 as a clinical target.
CONCLUSION
FGF23 is an endocrine hormone that uses αKlotho as a cofactor to exert its physiological effects on traditional, on-target organs, such as the kidney and parathyroid glands, thereby regulating phosphate homeostasis and mineral metabolism. In CKD patients, excess FGF23 also exerts αklotho-independent effects on nontraditional, off-target organs, such as the heart, liver, and cells of the immune system. These maladaptive effects result in LVH, inflammation, kidney disease progression, and impaired immune function. Clinically, FGF23 has relevant associations with cardiovascular morbidity and mortality in CKD. Phosphate binders, calcimimetics, and iron therapy have been proposed as possible therapeutic approaches. However, solid evidence regarding the effects of lowering FGF23 on improving outcomes is still missing. In addition, many other open questions should be addressed before FGF23 can be validated as a true clinical target in CKD.
FGF23 was identified less than 20 years ago and the next few years are likely to definitively reveal great progress in our understanding of FGF23 in both health and disease.
References
1. Park M, Hsu CY, Li Y, Mishra RK, Keane M, Rosas SE, et al. Associations between kidney function and subclinical cardiac abnormalities in CKD. J Am Soc Nephrol 2012; 23(10): 1725-1734. [ Links ]
2. Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med 2004; 351(13): 1296-1305. [ Links ]
3. Kendrick J, Chonchol MB. Nontraditional risk factors for cardiovascular disease in patients with chronic kidney disease. Nat Clin Pract Nephrol 2008; 4(12): 672-681. [ Links ]
4. Agarwal R. The challenge of discovering patient-level cardiovascular risk factors in chronic kidney disease. Kidney Int 2008; 73(12): 1340-1342. [ Links ]
5. Hu MC, Shiizaki K, Kuro-o M, Moe OW. Fibroblast growth factor 23 and Klotho: physiology and pathophysiology of an endocrine network of mineral metabolism. Annu Rev Physiol 2013; 75: 503-533. [ Links ]
6. Itoh N, Ornitz DM. Fibroblast growth factors: from molecular evolution to roles in development, metabolism and disease. J Biochem 2011; 149(2): 121-130. [ Links ]
7. Martin A, David V, Quarles LD. Regulation and function of the FGF23/klotho endocrine pathways. Physiol Rev 2012; 92(1): 131-155. [ Links ]
8. Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 2006; 444(7120): 770-774. [ Links ]
9. Liu S, Quarles LD. How fibroblast growth factor 23 works. J Am Soc Nephrol 2007; 18(6): 1637-1647. [ Links ]
10. Wolf M. Update on fibroblast growth factor 23 in chronic kidney disease. Kidney Int 2012; 82(7): 737-747. [ Links ]
11. Prie D, Urena TP, Friedlander G. Latest findings in phosphate homeostasis. Kidney Int 2009; 75(9): 882-889. [ Links ]
12. Shimada T, Urakawa I, Yamazaki Y, Hasegawa H, Hino R, Yoneya T, et al. FGF-23 transgenic mice demonstrate hypophosphatemic rickets with reduced expression of sodium phosphate cotransporter type IIa. Biochem Biophys Res Commun 2004; 314(2): 409-414. [ Links ]
13. Erben RG. Pleiotropic actions of FGF23. Toxicol Pathol 2017; 45(7): 904-910. [ Links ]
14. Gattineni J, Bates C, Twombley K, Dwarakanath V, Robinson ML, Goetz R, et al. FGF23 decreases renal NaPi-2a and NaPi-2c expression and induces hypophosphatemia in vivo predominantly via FGF receptor 1. Am J Physiol Renal Physiol 2009; 297(2): F282-F291. [ Links ]
15. Hasegawa H, Nagano N, Urakawa I, Yamazaki Y, Iijima K, Fujita T, et al. Direct evidence for a causative role of FGF23 in the abnormal renal phosphate handling and vitamin D metabolism in rats with early-stage chronic kidney disease. Kidney Int 2010; 78(10): 975-980. [ Links ] [ Links ]
17. Isakova T, Wahl P, Vargas GS, Gutierrez OM, Scialla J, Xie H, et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int 2011; 79(12): 1370-1378. [ Links ]
18. Gutierrez O, Isakova T, Rhee E, Shah A, Holmes J, Collerone G, et al. Fibroblast growth factor-23 mitigates hyperphosphatemia but accentuates calcitriol deficiency in chronic kidney disease. J Am Soc Nephrol 2005; 16(7): 2205-2215. [ Links ]
19. Vervloet MG, Sezer S, Massy ZA, Johansson L, Cozzolino M, Fouque D. The role of phosphate in kidney disease. Nat Rev Nephrol 2017; 13(1): 27-38. [ Links ]
20. David V, Martin A, Isakova T, Spaulding C, Qi L, Ramirez V, et al. Inflammation and functional iron deficiency regulate fibroblast growth factor 23 production. Kidney Int 2016; 89(1): 135-146. [ Links ]
21. Mace ML, Gravesen E, Nordholm A, Hofman-Bang J, Secher T, Olgaard K, et al. Kidney fibroblast growth factor 23 does not contribute to elevation of its circulating levels in uremia. Kidney Int 2017; 92(1): 165-178. [ Links ]
22. Leifheit-Nestler M, Grosse SR, Flasbart K, Richter B, Kirchhoff F, Ziegler WH, et al. Induction of cardiac FGF23/FGFR4 expression is associated with left ventricular hypertrophy in patients with chronic kidney disease. Nephrol Dial Transplant 2016; 31(7): 1088-1099. [ Links ]
23. Masuda Y, Ohta H, Morita Y, Nakayama Y, Miyake A, Itoh N, et al. Expression of Fgf23 in activated dendritic cells and macrophages in response to immunological stimuli in mice. Biol Pharm Bull 2015; 38(5): 687-693. [ Links ]
24. Christov M, Waikar SS, Pereira RC, Havasi A, Leaf DE, Goltzman D, et al. Plasma FGF23 levels increase rapidly after acute kidney injury. Kidney Int 2013; 84(4): 776-785. [ Links ]
25. Toro L, Barrientos V, Leon P, Rojas M, Gonzalez M, Gonzalez-Ibanez A, et al. Erythropoietin induces bone marrow and plasma fibroblast growth factor 23 during acute kidney injury. Kidney Int 2018; doi:10.1016/j.kint.2017.11.018 [ Links ]
26. Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, et al. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci U S A 2001; 98(11): 6500-6505. [ Links ]
27. Benet-Pages A, Lorenz-Depiereux B, Zischka H, White KE, Econs MJ, Strom TM. FGF23 is processed by proprotein convertases but not by PHEX. Bone 2004; 35(2): 455-462. [ Links ]
28. Goetz R, Nakada Y, Hu MC, Kurosu H, Wang L, Nakatani T, et al. Isolated C-terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23-FGFR-Klotho complex formation. Proc Natl Acad Sci U S A 2010; 107(1): 407-412. [ Links ]
29. Smith ER, Cai MM, McMahon LP, Holt SG. Biological variability of plasma intact and C-terminal FGF23 measurements. J Clin Endocrinol Metab 2012; 97(9): 3357-3365. [ Links ]
30. Bhattacharyya N, Wiench M, Dumitrescu C, Connolly BM, Bugge TH, Patel HV, et al. Mechanism of FGF23 processing in fibrous dysplasia. J Bone Miner Res 2012; 27(5): 1132-1141. [ Links ]
31. Shimada T, Urakawa I, Isakova T, Yamazaki Y, Epstein M, Wesseling-Perry K, et al. Circulating fibroblast growth factor 23 in patients with end-stage renal disease treated by peritoneal dialysis is intact and biologically active. J Clin Endocrinol Metab 2010; 95(2):578-585. [ Links ]
32. Burnett SM, Gunawardene SC, Bringhurst FR, Juppner H, Lee H, Finkelstein JS. Regulation of C-terminal and intact FGF-23 by dietary phosphate in men and women. J Bone Miner Res 2006; 21(8): 1187-1196. [ Links ]
33. Bacchetta J, Cochat P, Salusky IB, Wesseling-Perry K. Uric acid and IGF1 as possible determinants of FGF23 metabolism in children with normal renal function. Pediatr Nephrol 2012; 27(7): 1131-1138. [ Links ]
34. Imel EA, Peacock M, Gray AK, Padgett LR, Hui SL, Econs MJ. Iron modifies plasma FGF23 differently in autosomal dominant hypophosphatemic rickets and healthy humans. J Clin Endocrinol Metab 2011; 96(11): 3541-3549. [ Links ]
35. Gutierrez OM, Mannstadt M, Isakova T, Rauh-Hain JA, Tamez H, Shah A, et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med 2008; 359(6): 584-592. [ Links ]
36. Fliser D, Kollerits B, Neyer U, Ankerst DP, Lhotta K, Lingenhel A, et al. Fibroblast growth factor 23 (FGF23) predicts progression of chronic kidney disease: the Mild to Moderate Kidney Disease (MMKD) Study. J Am Soc Nephrol 2007; 18(9): 2600-2608. [ Links ]
37. Larsson T, Olauson H. [A brief update on FGF23 for the clinical nephrologist]. G Ital Nefrol 2014; 31(2). [ Links ]
38. Faul C, Amaral AP, Oskouei B, Hu MC, Sloan A, Isakova T, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest 2011; 121(11): 4393-4408. [ Links ]
39. Di Marco GS, Reuter S, Kentrup D, Ting L, Ting L, Grabner A, et al. Cardioprotective effect of calcineurin inhibition in an animal model of renal disease. Eur Heart J 2011; 32(15): 1935-1945. [ Links ]
40. Grabner A, Amaral AP, Schramm K, Singh S, Sloan A, Yanucil C, et al. Activation of cardiac fibroblast growth factor receptor 4 causes left ventricular hypertrophy. Cell Metab 2015; 22(6): 1020-1032. [ Links ]
41. Di Marco GS, Reuter S, Kentrup D, Grabner A, Amaral AP, Fobker M, et al. Treatment of established left ventricular hypertrophy with fibroblast growth factor receptor blockade in an animal model of CKD. Nephrol Dial Transplant 2014; 29(11): 2028-2035. [ Links ]
42. Yanochko GM, Vitsky A, Heyen JR, Hirakawa B, Lam JL, May J, et al. Pan-FGFR inhibition leads to blockade of FGF23 signaling, soft tissue mineralization, and cardiovascular dysfunction. Toxicol Sci 2013; 135(2): 451-464. [ Links ]
43. Shalhoub V, Shatzen EM, Ward SC, Davis J, Stevens J, Bi V, et al. FGF23 neutralization improves chronic kidney disease-associated hyperparathyroidism yet increases mortality. J Clin Invest 2012; 122(7): 2543-2553. [ Links ]
44. Hagel M, Miduturu C, Sheets M, Rubin N, Weng W, Stransky N, et al. First selective small molecule inhibitor of FGFR4 for the treatment of hepatocellular carcinomas with an activated FGFR4 signaling pathway. Cancer Discov 2015; 5(4): 424-437. [ Links ]
45. Grabner A, Schramm K, Silswal N, Hendrix M, Yanucil C, Czaya B, et al. FGF23/FGFR4-mediated left ventricular hypertrophy is reversible. Sci Rep 2017; 7(1): 1993. [ Links ]
46. Singh S, Grabner A, Yanucil C, Schramm K, Czaya B, Krick S, et al. Fibroblast growth factor 23 directly targets hepatocytes to promote inflammation in chronic kidney disease. Kidney Int 2016; 90(5): 985-996. [ Links ]
47. Han X, Li L, Yang J, King G, Xiao Z, Quarles LD. Counter-regulatory paracrine actions of FGF-23 and 1,25(OH)2 D in macrophages. FEBS Lett 2016; 590(1): 53-67. [ Links ]
48. Dai B, David V, Martin A, Huang J, Li H, Jiao Y, et al. A comparative transcriptome analysis identifying FGF23 regulated genes in the kidney of a mouse CKD model. PLoS One 2012; 7(9): e44161. [ Links ]
49. Rossaint J, Oehmichen J, Van AH, Reuter S, Pavenstadt HJ, Meersch M, et al. FGF23 signaling impairs neutrophil recruitment and host defense during CKD. J Clin Invest 2016; 126(3): 962-974. [ Links ]
50. Rossaint J, Unruh M, Zarbock A. Fibroblast growth factor 23 actions in inflammation: a key factor in CKD outcomes. Nephrol Dial Transplant 2017; 32(9): 1448-1453. [ Links ]
51. Hanudel M, Juppner H, Salusky IB. Fibroblast growth factor 23: fueling the fire. Kidney Int 2016; 90(5): 928-930. [ Links ]
52. Francis C, David V. Inflammation regulates fibroblast growth factor 23 production. Curr Opin Nephrol Hypertens 2016; 25(4): 325-332. [ Links ]
53. Wolf M, Koch TA, Bregman DB. Effects of iron deficiency anemia and its treatment on fibroblast growth factor 23 and phosphate homeostasis in women. J Bone Miner Res 2013; 28(8): 1793-1803. [ Links ]
54. Silverstein DM. Inflammation in chronic kidney disease: role in the progression of renal and cardiovascular disease. Pediatr Nephrol 2009; 24(8): 1445-1452. [ Links ]
55. Munoz MJ, Isakova T, Ricardo AC, Xie H, Navaneethan SD, Anderson AH, et al. Fibroblast growth factor 23 and Inflammation in CKD. Clin J Am Soc Nephrol 2012; 7(7): 1155-1162. [ Links ]
56. Manghat P, Fraser WD, Wierzbicki AS, Fogelman I, Goldsmith DJ, Hampson G. Fibroblast growth factor-23 is associated with C-reactive protein, serum phosphate and bone mineral density in chronic kidney disease. Osteoporos Int 2010; 21(11): 1853-1861. [ Links ]
57. Isakova T, Xie H, Yang W, Xie D, Anderson AH, Scialla J, et al. Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. JAMA 2011; 305(23): 2432-2439. [ Links ]
58. Jean G, Terrat JC, Vanel T, Hurot JM, Lorriaux C, Mayor B, et al. High levels of serum fibroblast growth factor (FGF)-23 are associated with increased mortality in long haemodialysis patients. Nephrol Dial Transplant 2009; 24(9): 2792-2796. [ Links ]
59. Parker BD, Schurgers LJ, Brandenburg VM, Christenson RH, Vermeer C, Ketteler M, et al. The associations of fibroblast growth factor 23 and uncarboxylated matrix Gla protein with mortality in coronary artery disease: the Heart and Soul Study. Ann Intern Med 2010; 152(10): 640-648. [ Links ]
60. Gutierrez OM, Januzzi JL, Isakova T, Laliberte K, Smith K, Collerone G, et al. Fibroblast growth factor 23 and left ventricular hypertrophy in chronic kidney disease. Circulation 2009; 119(19): 2545-2552. [ Links ]
61. Mirza MA, Larsson A, Melhus H, Lind L, Larsson TE. Serum intact FGF23 associate with left ventricular mass, hypertrophy and geometry in an elderly population. Atherosclerosis 2009; 207(2): 546-551. [ Links ]
62. Ix JH, Katz R, Kestenbaum BR, de Boer IH, Chonchol M, Mukamal KJ, et al. Fibroblast growth factor-23 and death, heart failure, and cardiovascular events in community-living individuals: CHS (Cardiovascular Health Study). J Am Coll Cardiol 2012; 60(3): 200-207. [ Links ]
63. Scialla JJ, Xie H, Rahman M, Anderson AH, Isakova T, Ojo A, et al. Fibroblast growth factor-23 and cardiovascular events in CKD. J Am Soc Nephrol 2014; 25(2):349-360. [ Links ]
64. Isakova T, Cai X, Lee J, Xie D, Wang X, Mehta R, et al. Longitudinal FGF23 trajectories and mortality in patients with CKD. J Am Soc Nephrol 2018; 29(2): 579-590. [ Links ]
65. Leifheit-Nestler M, Grabner A, Hermann L, Richter B, Schmitz K, Fischer DC, et al. Vitamin D treatment attenuates cardiac FGF23/FGFR4 signaling and hypertrophy in uremic rats. Nephrol Dial Transplant 2017; 32(9): 1493-1503. [ Links ]
66. Wetmore JB, Liu S, Krebill R, Menard R, Quarles LD. Effects of cinacalcet and concurrent low-dose vitamin D on FGF23 levels in ESRD. Clin J Am Soc Nephrol 2010; 5(1): 110-116. [ Links ]
67. Moe SM, Chertow GM, Parfrey PS, Kubo Y, Block GA, Correa-Rotter R, et al. Cinacalcet, fibroblast growth Factor-23, and cardiovascular disease in hemodialysis: the Evaluation of cinacalcet HCl therapy to lower cardiovascular events (EVOLVE) trial. Circulation 2015; 132(1): 27-39. [ Links ]
68. Kim HJ, Kim H, Shin N, Na KY, Kim YL, Kim D, et al. Cinacalcet lowering of serum fibroblast growth factor-23 concentration may be independent from serum Ca, P, PTH and dose of active vitamin D in peritoneal dialysis patients: a randomized controlled study. BMC Nephrol 2013; 14: 112. [ Links ]
69. Adema AY, de Jong MA, de Borst MH, ter Wee PM, Vervloet MG. Phosphate binding therapy to lower serum fibroblast-growth-factor-23 concentrations in chronic kidney disease: rationale and study design of the sevelamer on FGF23 trial (SoFT). Nephron 2016; 134(4): 215-220. [ Links ]
70. Soriano S, Ojeda R, Rodriguez M, Almaden Y, Rodriguez M, Martin-Malo A, et al. The effect of phosphate binders, calcium and lanthanum carbonate on FGF23 levels in chronic kidney disease patients. Clin Nephrol 2013; 80(1): 17-22. [ Links ]
71. Liabeuf S, Ryckelynck JP, El EN, Urena P, Combe C, Dussol B, et al. Randomized clinical trial of sevelamer carbonate on serum klotho and fibroblast growth factor 23 in CKD. Clin J Am Soc Nephrol 2017; 12(12): 1930-1940. [ Links ]
72. Iguchi A, Kazama JJ, Yamamoto S, Yoshita K, Watanabe Y, Iino N, et al. Administration of ferric citrate hydrate decreases circulating FGF23 levels independently of serum phosphate levels in hemodialysis patients with iron deficiency. Nephron 2015; 131(3): 161-166. [ Links ]
Giovana S. Di Marco
Albert-Schweitzer-Campus 1, Gebäude A14, 48149 Münster, Germany.
Email: dimarco@uni-muenster.de
Disclosure of potential conflicts of interest: none declared.
Received for publication: Mar 8, 2018 Accepted in revised form: Mar 16, 2018

