










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
versão impressa ISSN 0872-0169
Port J Nephrol Hypert vol.32 no.1 Lisboa mar. 2018
CASE REPORT
C3 glomerulonephritis disguised as Postinfectious GN
Alice Lança, Paulo Santos, Karina Lopes, Francisco Ferrer, Ana Vila Lobos
Nephrology Department, Centro Hospitalar Médio Tejo, Torres Novas, Portugal
ABSTRACT
It is not uncommon for atypical cases of postinfectious glomerulonephritis (PIGN) to be confused with C3 glomerulonephritis (C3GN) due to considerable overlap of their clinical and histopathological features. We present a case of a 42-year-old man who was hospitalized with hematuria and worsening renal function after an episode of pharyngitis. Following a complete negative serological examination, the indolent clinical course associated with the findings of diffuse proliferative and exudative glomerulonephritis with dominant C3 staining and subendothelial and mesangial electron-dense deposits on the kidney biopsy were fundamental for the final diagnosis. The genetic test identified two mutations involved in the alternative complement pathway. Steroids and mycophenolate mofetil were initiated with improved renal function. This case alerts us that the presence of atypical clinical or histological features of apparent PIGN should raise suspicion of C3 glomerulopathy (C3G) because only a correct diagnosis will allow adequate treatment and a better long-term prognosis.
Keywords: C3 glomerulonephritis; Chronic Kidney Disease; Postinfectious glomerulonephritis
BACKGROUND
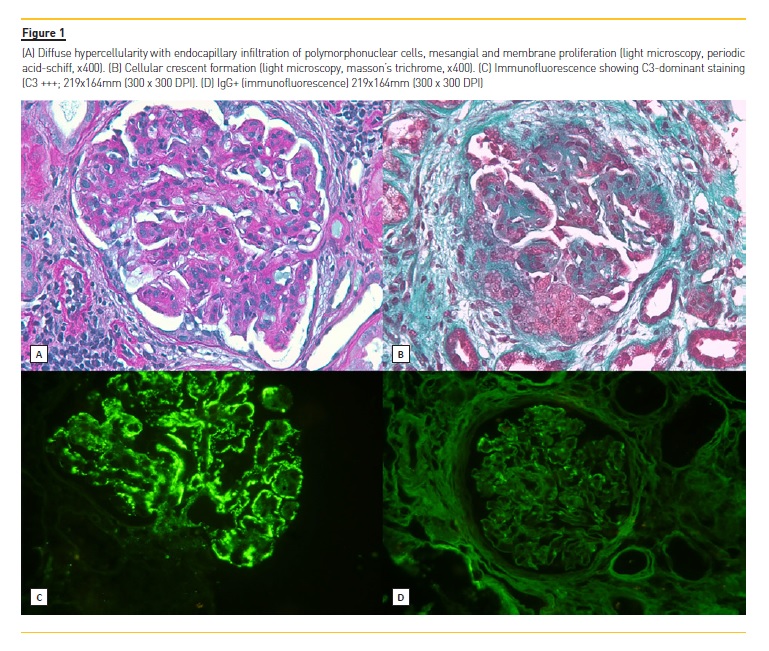
Since the last century, acute nephritic syndrome preceded by an infection has been known as postinfectious glomerulonephritis (PIGN)1. It classically results from an immune-complex mediated GN presenting with hematuria, proteinuria, hypertension, edema and renal function impairment of variable degree and C3 consumption that often resolves over the next 6–8 weeks2. The most common histological finding is a proliferative glomerulonephritis with diffuse hypercellularity and endocapillary infiltration of neutrophils (exudative GN) with both immunoglobulin (IgG) and C3 deposition on immunofluorescence (IMF). This pattern suggests activation of complement by antibodydependent pathways. However, recent advances in the understanding of complement-mediated kidney injury have led to the identification of new pathological entities, such as C3 glomerulopathy (C3G). C3N is a rare abnormality (1 to 2 per million) characterized by prominent glomerular accumulation of C3 with minimal or absent immunoglobulin deposition secondary to alternative complement pathway dysregulation3. The differential diagnosis with PIGN can be a real challenge since it may have a very similar clinical presentation. In this report, we describe a male patient who presented with nephritic syndrome after an episode of sore throat and whose clinical and pathological findings were initially consistent with PIGN. The indolent clinical course together with the co-dominance of C3 in the glomeruli associated with the ultrastructural findings were determinant for the final diagnosis.
CASE REPORT
A 42-year-old Caucasian man with a prior history of migraine was admitted to the Nephrology Department with worsening renal function. The patient had been well until approximately 3 weeks before admission when malaise, fatigue and anorexia developed. He initially attributed these symptoms to an episode of pharyngitis but during this time he also developed dyspnea, foamy urine and peripheral edema with weight gain (7kg). Before admission the patient was on no medications. On examination he was afebrile with a blood pressure of 220/100mmHg. He had periorbital edema and moderate (2+) peripheral pitting edema.
Oropharynx showed no changes. The remainder of the physical examination was unremarkable. Laboratory testing revealed a hemoglobin level of 12.7 g/dL, a 16400 leucocyte count, negative C-reactive protein (CRP), creatinine 2.7 mg/dL, urea 72 mg/dL and serum albumin 3.1 g/dL. The platelet count, red-cell indexes, coagulation and liver-function tests were normal as were blood levels of electrolytes and glucose. Urinalysis revealed a moderate amount of occult blood (hb: 0.20 mg/dL) and a protein of 250 mg per deciliter (3+). On microscopic examination, there were 67 red cells and 6 white cells per high-power field. Protein/creatinine ratio was 4224 mg/g. Kidney ultrasound showed normal-sized kidneys with normal parenchyma and no signs of lithiasis or obstructive uropathy. The serological workup (ANA, ANCA, anti-GBM and complement) was negative. Anti-streptolysin O (ASOT), serum and urinary electrophoresis as well as immunofixation were normal.
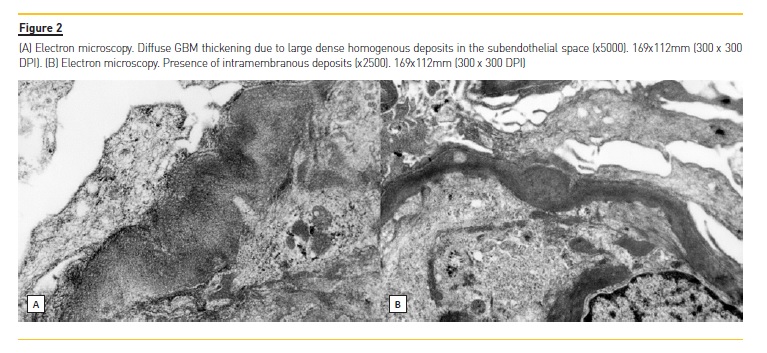
In order to clarify the clinical picture, a kidney biopsy was performed; a diffuse proliferative and exudative pattern of glomerulonephritis notable for the presence of infiltrating neutrophils with C3-dominant staining by IMF (Figure 1, was present. Based on the biopsy findings and clinical history, acute PIGN appeared to be the most likely diagnosis. Blood and urinary cultures, and imaging tests (transthoracic echocardiogram, abdominal and renal ultrasonography (US) and thoracoabdominal-pelvic computed tomography (CT) without contrast) were performed and none showed major alterations. Meanwhile, supportive measures and antimicrobial therapy were initiated, however, renal function continued to deteriorate (creatinine up to 4.8 mg/dL) and both macroscopic hematuria (92 erythrocytes per high power field) and nephrotic range proteinuria (6100 mg/24h) persisted. After excluding an infectious process, methylprednisolone was administered for 3 days (500mg/day) followed by oral prednisolone 60mg/day (<1mg/kg/day) for 3 months. A mild recovery of renal function (creatinine: 3.8 mg/dL) and a proteinuria reduction was observed but hematuria and peripheral edema persisted. A trial of mycophenolate mofetil (MMF) was then offered (initially 2.5 g/ day followed by 1.5 g/day due to gastrointestinal side effects) and prednisolone was slowly reduced with significant improvement of hematoproteinuria. Simultaneously, genetic study and electron microscopy (EM) evaluation were requested. Two mutations were detected: a variant mutation of uncertain significance (c.94A>C [p.Lys32Gln]) in the complement factor I (CFI) gene and a mutation with predicted deleterious nonsynonymous rare variants (c.467G>A [p.Cys156Tyr]) in the MASP2 gene. On EM, the localization and the characteristics of the electron-dense deposits (subendothelial and mesangial) made the distinction between C3 glomerulonehpritis (C3GN) and Dense Deposit Disease (DDD), suggesting C3GN as the final diagnosis (Figure 2). Ten months after the initiation of MMF, the patients had 24-hour urine protein excretion of 60 mg/day, a creatinine level of 1.9 mg/dL, estimated glomerular filtration rate of 43 mL/min/1.73 m2 (CKD-EPI) with great improvement in health status.
DISCUSSION
Here, we report a case of a previously healthy 42-year-old man who presented at the emergency department with arterial hypertension, acute kidney failure (AKI), hematuria and nephrotic range proteinuria following an episode of pharyngitis. Routine investigations suggested primary glomerulonephritis and kidney biopsy revealed morphological features consistent with proliferative GN.
Nephritic syndrome, a glomerular inflammation resulting in a reduction in glomerular filtration rate (GFR), non-nephrotic pro-teinuria, edema, hypertension and hematuria with RBC casts, is classically seen in PIGN. PIGN is an immunologically mediated glomerular injury triggered by an infection with C3 consumption that often resolves over the next 6–8 weeks2.
C3G is a rare, recently described clinical entity characterized by predominant glomerular C3 deposition and electron-dense deposits that may be preceded by an upper respiratory tract infection and mimic PIGN4.
Given the similar clinical presentation, its not uncommon for C3G to be misdiagnosed as PIGN, particularly in those with persistent and recurrent disease with hypocomplementemia and high titers of ASOT5. In some cases, the dis-tinction from C3GN may only be possible by following up the patient to see if resolution of the disease occurs.
In our patient, the diagnosis of PIGN was favoured initially based on the clinical presentation of AKI preceded by an inciting infectious event (pharyngitis) the presence of leukocytosis and the pattern of proliferative endocapillary exudative GN6. On the other hand, many factors pointed in another direction. The patient was a previously healthy adult, living in a developed country with no signs of fever or infective agent, normal serum C3 and C4 levels, normal CRP, negative ASOT, negative cultures and inconclusive imaging. Additionally, and very important to mention, the lack of clinical improvement with persistent active urine sediment and deterioration of renal function following the administration of broad-spectrum antibiotics made PIGN less likely7.
PIGN usually resolves spontane-ously over a few days and has a favorable prognosis. Antibiotic treatment given for 7 to 10 days often leads to full recovery. Rarely, mild proteinuria (<500 mg/day) and microscopic hematuria may persist for up to 1 year without worsening the long-term prognosis. Nevertheless, some individuals, especially adults, may have persistent impaired renal function, proteinuria, or hyper-tension, suggesting that PIGN could be a self-limiting form of C3G or that C3G may be initiated by a streptococcal infection.
Some atypical PIGN may be related with mutations in complement-regulating proteins and with autoantibodies to the C3 convertase, others are in fact, C3G8.
C3G which encompasses both DDD and C3GN, does not appear to have such a benign course. In C3GN, infection itself can act as a tickover of the alternative complement pathway (AP) which is usually active at a low level and is tightly controlled, resulting in excessive production of C3 and deleterious effects1,9. Multiple complement regulators and inhibitors operate at every level to prevent this self-mediated damage. However, acquired conditions such as C3 nephritic factor (C3NeF) antibody and genetic alterations in complement proteins (mutations in serum factor H, complement factor H-related (CFHR), factor B, I and membrane cofactor protein, MCP) often exacerbate complement-mediated damage and tip the balance from protection to destruction10.
Histologically, C3G may exhibit a myriad of patterns such as membranoproliferative GN (MPGN), mesangial proliferative GN, endocapillary proliferative (sometimes exudative) and crescentic GN6 but the diagnosis is relatively straightforward if there is isolated C3 deposition with typical deposits on EM. Until quite recently, the main focus was the classical pathway which was triggered by circulating immune complexes as a consequence of persistent antigenemia causing intense C3 and IgG deposition along the capillary walls and in the mesangium. However, the absence of IgG deposits in the presence of complement products or the dominance of C3 over IgG (at least two orders of magnitude less) points to an activation of the AP11.
On kidney biopsy, our patient showed a strong codominance of C3 staining in both mesangium and capillary walls and the presence of electron-dense subendothelial and mesangial deposits on the EM5. Based on the biopsy findings along with the clinical evolution, the most probable diagnosis was C3GN. In order to investigate the cause of this entity, a genetic screening was requested.
A variant mutation (c.94A>C [p.Lys32Gln]) in the complement factor I (CFI), a regulating gene of AP, was identified; however to the best of our knowledge, of uncertain significance. Another mutation (c.467G>A [p.Cys156Tyr]) in the MASP2 gene was detected and unlike the former it has been previously described in the literature (Bu et al) and in several databases such as dbSNP, gnomAP, ESP and GoNL as pathogenic (predicted deleterious nonsynonymous rare variant) involved in some complement disorders12. Unfortunately, for technical reasons, C3 nephritic factor was not requested. For this reason, we cannot confirm the pathophysiological mechanism involved in this disease.
The optimal treatment for C3 glomerulopathy remains undefined. Despite the increasing recognition of this disease in recent decades, the paucity of data on clinical and pathological features and its association with disease outcomes remain scarce. The lack of randomized trials to provide information on treatment decisions has led to low-quality evidence-based treatment. Currently, the therapeutic options include fresh frozen plasma (for genetic deficiencies), plasma exchange, immunosuppression (glucocorticoids, cyclophosphamide and mycophenolate mofetil), rituximab and eculizumab if it is autoantibody-related. To date, targeted therapy to C3 glomerulopathy has not yet been defined, although disease-specific therapeutic agents that modulate complement pathways are being developed13. Renal transplantation should be considered in patients with C3 GN, despite the rate of recurrence in the allograft6.
Data suggests that C3 GN prognosis appears to be variable, mostly poor and worse than for PIGN. Therefore, the presence of any atypical clinical or histological features in a case of apparent PIGN should raise suspicion of a C3 glomerulopathy and prompt immediate characterization of this group of diseases in order not to delay the final diagnosis. Since the awareness of the diagnostic criteria has recently increased, there will likely be an increase in patients diagnosed with C3GP which will help define the histological predictors of renal outcome.
References
1. Khalighi MA, Wang S, Henriksen KJ, Bock M, Keswani M, Meehan SM, et al. Revisiting post-infectious glomerulonephritis in the emerging era of C3 glomerulopathy. Clin Kidney J. 2016;9(3):397–402. [ Links ]
2. Kanjanabuch T, Kittikowit W, Eiam-Ong S. An update on acute postinfectious glomerulonephritis worldwide. Nat Rev Nephrol [Internet]. 2009;5(5):259–69. [ Links ]
3. Sethi S, Fervenza FC, Zhang Y, Zand L, Vrana JA, Nasr SH, et al. C3 glomerulonephritis: clinicopathological findings, complement abnormalities, glomerular proteomic profile, treatment, and follow-up. Kidney Int [Internet]. 2012;82(4):465–73. [ Links ]
4. Pickering M, Cook HT. Complement and glomerular disease: new insights. Curr Opin Nephrol Hypertens. 2011;20(3):271–7. [ Links ]
5. Medjeral-Thomas NR, OShaughnessy MM, ORegan JA, Traynor C, Flanagan M, Wong L, et al. C3 glomerulopathy: Clinicopathologic features and predictors of outcome. Clin J Am Soc Nephrol. 2014;9(1):46–53. [ Links ]
6. Cook HT, Pickering MC. Histopathology of MPGN and C3 glomerulopathies. Nat Rev Nephrol [Internet]. 2014;11(1):14–22. [ Links ]
7. Stratta P, Musetti C, Barreca A, Mazzucco G. New trends of an old disease: The acute post infectious glomerulonephritis at the beginning of the new millenium. J Nephrol. 2014;27(3):229–39. [ Links ]
8. Ito N, Ohashi R, Nagata M. C3 glomerulopathy and current dilemmas. Clin Exp Nephrol. 2017;21(4):541–51. [ Links ]
9. Barbour TD, Ruseva MM, Pickering MC. Update on C3 glomerulopathy. Nephrol Dial Transplant. 2016;31(5):717–25. [ Links ]
10. Bomback AS, Markowitz GS, Appel GB. Complement-mediated glomerular diseases: A tale of 3 pathways. Kidney Int Reports [Internet]. 2016;1(3):148–55. [ Links ]
11. Sethi S, Fervenza FC. Membranoproliferative glomerulonephritis: Pathogenetic heterogeneity and proposal for a new classification. Semin Nephrol [Internet]. 2011;31(4):341–8. [ Links ]
12. Bu F, Maga T, Meyer NC, Wang K, Thomas CP, Nester CM, et al. Comprehensive genetic analysis of complement and coagulation genes in atypical hemolytic uremic syndrome. J Am Soc Nephrol [Internet]. 2014;25(1):55–64. [ Links ]
13. Bomback AS. Anti-complement therapy for glomerular diseases. Adv Chronic Kidney Dis [Internet]. 2014;21(2):152–8. [ Links ]
Alice Lança
Department of Nephrology, Centro Hospitalar Médio Tejo
Av. Xanana Gusmão, s/n
2350-754 Torres Novas, Portugal
Email: alicelancabaptista@gmail.com
ACKNOWLEDGMENTS
The authors would like to thank both Dr. Maria Fernanda Carvalho and Dr. Mário Góis (Kidney Morphology Laboratory, Department of Nephrology, Hospital Curry Cabral, Lisbon, Portugal) for interpreting this renal biopsy and suggesting the final diagnosis that led to the appropriate therapeutic strategy while, at the same time, providing key prognostic information.
Disclosure of potential conflicts of interest: none declared
Received for publication: Dec 2, 2017
Accepted in revised form: Dec 15, 2017


{kind=link}
{kind=link}