










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
Print version ISSN 0872-0169
Port J Nephrol Hypert vol.32 no.3 Lisboa Sept. 2018
REVIEW ARTICLE
Kidney diseases with ocular involvement: a systematic review
Rachele Escoli1, Ricardo Oliveira2, Paulo Santos1, Ana Vila Lobos1
1 Nephrology department, Centro Hospitalar do Médio Tejo, Torres Novas, Portugal
2 Ophthalmology department, Centro Hospitalar do Médio Tejo, Tomar, Portugal
ABSTRACT
Chronic kidney disease is an emerging health problem worldwide. The eye shares striking structural, developmental, and genetic pathways with the kidney, suggesting that kidney and ocular disease may be closely linked. The aim of this paper, beyond exploring the underlying pathogenic mechanisms and common risk factors, is a review of the main diseases with ocular and renal involvement.
Key Words: Chronic kidney disease, nephropathy, ophthalmopathy, retinopathy, visual loss
INTRODUCTION
How can findings in the eye prompt a diagnosis of a renal disease? And vice versa, how can the diagnosis of renal disease lead to an ophthalmology consultation to rule out eye disease? A study on patients with chronic kidney disease (CKD) revealed retinal pathologies in up to 25% of patients1. Hypertensive and diabetic diseases are by far the leading pathologies, but other entities can also be found and will be discussed1.
The kidney and eye share striking structural, developmental, physiological, and pathogenic pathways. For example, both the glomerulus and choroid have extensive vascular networks of similar structure. The inner retina and glomerular filtration barrier share similar developmental pathways, and the renin–angiotensin–aldosterone hormonal cascade is found in both the eye and the kidney2.
This review aims to analyze common risk factors and pathogenic mechanisms underlying CKD and eye diseases and to review the main diseases which have ocular and renal involvement, organized as follows: systemic, vascular, autoimmune, infectious, genetic and related to kidney transplantation.
RISK FACTORS AND PATHOPHYSIOLOGICAL MECHANISMS OF CKD AND EYE DISEASES
CKD is an emerging health problem worldwide associated with serious cardiovascular and renal outcomes and decreased quality of life. More than 25 million adults in the United States are estimated to have CKD and over half a million to have end‑stage renal disease (ESRD) and CKD is expected to increase further with an aging population and with the increasing prevalence of risk factors, such as old age, smoking, hypertension, diabetes mellitus and obesity. CKD shares traditional cardiovascular risk factors and is increasingly recognized as a risk factor itself for cardiovascular disease (CVD). Some of these are also risk factors for ophthalmologic diseases2.
The main pathophysiological mechanisms that contribute to CKD are atherosclerosis, vascular remodeling, endothelial dysfunction, inflammation, and oxidative stress. These mechanisms are also implicated in many ocular diseases2 (Figure 1).
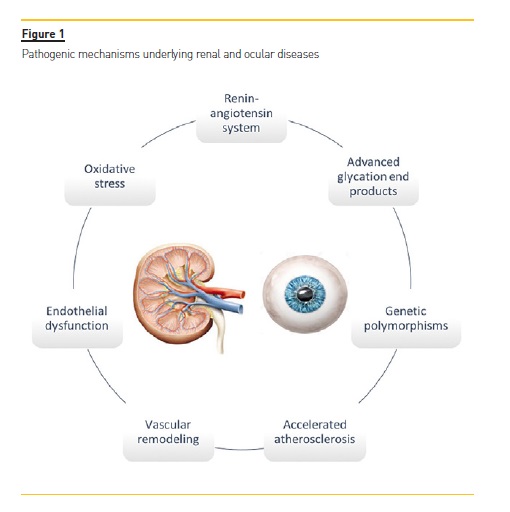
DISEASES WITH OCULAR AND RENAL INVOLVEMENT
There are several diseases with ocular and renal involvement. The review of diseases with involvement of the two organs will be made based on the following division: systemic, vascular, autoimmune, infectious, genetic and related to kidney transplantation.
Systemic disorders
Diabetes Mellitus
Diabetes mellitus (DM) is the primary cause of CKD and accounts for 40% of all new cases of ESRD recorded annually, but has multiorgan involvement3. Diabetic nephropathy (DN) and retinopathy are arguably the two most dreaded complications of diabetes and together they contribute to serious morbidity and mortality. Their progression can lead to ESRD and blindness4.
DN is characterized by persistent albuminuria, progressive decline of glomerular filtration rate, and elevation of blood pressure5,6. In patients with DN, the presence of albumin in urine not only signifies glomerular injury, but also reflects systemic endothelial abnormalities and vasculopathy, which can represent an independent risk factor for cardiovascular disease. Risk factors for DN include prolonged duration of diabetes, poor glycemic control, and hypertension3.
Patients with diabetes often develop ophthalmic complications, such as corneal abnormalities, glaucoma, iris neovascularization, cataracts, and neuropathies.
The most common and potentially blinding of these complications, however, is diabetic retinopathy (DR), which is, in fact, the leading cause of acquired blindness in aged persons7. The exact mechanism by which diabetes causes retinopathy remains unclear, but several theories have been postulated to explain the typical course and history of the disease8. In DR, chronic hyperglycemia causes endothelial damage, loss of pericytes, basement‑membrane thickening, breakdown of the blood‑retinal barrier, platelet aggregation, and leukocyte adhesion in retinal capillaries. The microstructure disarrangement and microcirculation dysfunction lead to vascular hyperpermeability and microaneurysm formation, as observed in nonproliferative DR. Excessive vascular leakage of fluids, proteins, or lipids in the macular area leads to the development of diabetic macular edema. As the disease progresses, capillaries close and arterioles become atrophied, and this matches the nonperfusion areas detected in patients fluorescein angiography. Eventually, chronic hypoxia induces the expression of several angiogenic growth factors, which results in retinal neovascularization, as observed in proliferative DR3.
In DN, chronic hyperglycemia also alters the expression of growth factors and cytokines in renal glomeruli, and these changes, in turn, result in an imbalance of the hemodynamics in glomerular cells. In the early stages, glomerular hypertrophy and hyperfiltration occur as glomeruli respond to the expression of hyperglycemia.
However, increased intraglomerular pressure and increased shear stress following loss of heparin sulfates in the glomeruli eventually lead to the thickening of the glomerular and tubular basement membrane, accumulation of the mesangial matrix, and albuminuria9‑11.
The correlation between diabetic retinopathy and diabetic nephropathy is so strong (a nearly 100% coincidence) that diabetes should be in doubt as the cause of renal insufficiency in any patient with a normal retinal exam12.
Vascular disorders
Hypertension
Hypertension is a major promoter of the decline in glomerular filtration rate (GFR) in both diabetic and nondiabetic kidney disease13. Large, observational, prospective trials in the general population showed that hypertension is a strong independent risk factor for ESRD. A strong relationship was observed between both systolic (SBP) and diastolic blood pressure (DBP) and ESRD, regardless of other known risk factors, in men who were recruited in the Multiple Risk Factor Intervention Trial. The relative risk for ESRD was >20‑fold higher for patients with stage 4 hypertension (SBP > 210 mmHg or DBP > 120 mmHg) than for patients with optimal BP levels (SBP < 120 mmHg and DBP < 80 mmHg). The study by the Okinawa General Health Maintenance Association confirmed these results in women as well14,15.
Hypertension‑related mechanisms that are involved in the progression of renal damage include the systemic BP load, the degree to which it is transmitted to the renal microvasculature (i.e., renal autoregulation), and local susceptibility factors to barotrauma, which is the degree of damage for any degree of BP load. Among these last factors, proteinuria, glomerular hypertrophy, fibrogenic mediators, genetic factors, and age are the most important16.
Along with causing heart and kidney problems, untreated high blood pressure can also affect eyesight and lead to eye disease. The most common ocular diseases directly related to hypertension are progressively increasing retinal microvascular changes, which are subsumed under the name hypertensive retinopathy.
Classically, the features are divided into four degrees, and their morphological classification has been widely used17. However, a more pathophysiological division has been proposed. This three‑degree classification includes mild – retinal arteriolar narrowing related to vasospasm, arteriolar wall thickening or opacification, and arteriovenous nicking, referred to as nipping, moderate – hemorrhages, either flame or dot‑shaped, cotton‑wool spots, hard exudates, and microaneurysms and severe – some or all of the above, plus optic disc edema. The presence of papilledema requires rapid lowering of the blood pressure18.
Autoimmune diseases
Sarcoidosis
Sarcoidosis is a multisystem granulomatous disorder of unknown etiology is characterized pathologically by the presence of noncaseous granulomas in involved organs. It typically affects young adults and initially presents with one or more of the following abnormalities: bilateral hilar adenopathy, pulmonary reticular opacities, skin, joint, and/or eye lesion. However, sarcoidosis can involve all organ systems to a varying extent and degree19.
In the eye, sarcoidosis can have intra‑ or extraocular manifestations. Intraocular manifestations are classified as anterior, intermediate, and/or posterior uveitis20.
Anterior uveitis typically causes pain, endothelial granulomas, and redness primarily at the limbus. Posterior or intermediate uveitis are more likely to be painless but more often associated with floaters. Visual loss may occur with anterior, intermediate, or posterior involvement. As ocular involvement may be asymptomatic, all patients should undergo ophthalmologic examination20. Based on consensus guidelines, the certainty of a diagnosis of ocular sarcoidosis is based upon the combination of intraocular findings and systemic evidence of sarcoidosis21. Extraocular orbital sarcoidosis can affect lacrimal glands, conjunctiva, extraocular muscles, and optic sheath, and may present as a soft tissue orbital mass.22.
In the kidney, clinically renal involvement occasionally occurs in sarcoidosis. Renal manifestations include abnormal calcium metabolism, nephrolithiasis and nephrocalcinosis, and acute interstitial nephritis with or without granuloma formation. The classic renal lesion is noncaseous granulomatous interstitial nephritis.
However, this lesion rarely causes clinically significant renal disease. Hypercalciuria and hypercalcemia are most often responsible for clinically significant renal disease. Glomerular disease, obstructive uropathy, and ESRD may also occur but are uncommon23. Several studies have examined the relative prevalence of the various renal lesions associated with sarcoidosis: nephrocalcinosis is estimated to occur in 5 percent of patients with sarcoidosis and may be the most common cause of chronic kidney disease (CKD) in sarcoidosis24.
Nephrolithiasis occurs in approximately 1 to 14 percent of patients with sarcoidosis. Interstitial nephritis with granuloma formation occurs in approximately 20 percent of patients. However, renal insufficiency is not always present25. Glomerular involvement is rare. A variety of different lesions have been described in isolated cases, including membranous nephropathy, IgA nephropathy, minimal change disease, a proliferative or crescentic glomerulonephritis, and focal segmental glomerulosclerosis23. Urinary tract obstruction is veryrare among patients with sarcoidosis24.
Tubulointerstitial Nephritis and Uveitis Syndrome
Tubulointerstitial nephritis and uveitis syndrome (TINU) describes a rare form of bilateral non‑granulomatous anterior uveitis found in a sub‑population of patients with tubulointerstitial nephritis (TIN). The uveitis is usually mild and the nephritis self‑limited. However, cases of chronic uveitis and renal failure have been reported26.
Although the cause of TINU is unknown, research has revealed various associations. Certain HLA genotypes (including HLA‑DQA1* 01:04 and DRB1*14) increase the relative risk of developing TINU in certain populations.
Medications have also been implicated (e.g. antibiotics or nonsteroidal anti‑inflammatory drugs)26.
In over 80% of cases, uveitis is bilateral, acute in onset, non‑granulomatous and affects only the anterior segment27.
However, there have been case reports of posterior uveitis and panuveitis (26, 27). Patients will usually present with typical anterior uveitis symptoms (eye pain, redness, decreased vision, and photophobia).
Renal manifestations with TINU syndrome are typical for acute interstitial nephritis. These may include flank pain, sterile pyuria, hematuria, proteinuria (usually subnephrotic range), renal insufficiency, and acute renal failure (ARF). Multiple proximal and distal tubular defects can be commonly seen, resulting in aminoaciduria, glucosuria, phosphaturia, and acidification defects. Renal sonography can demonstrate marked swelling of the kidneys28.
Systemic Lupus Erythematosus
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease that can affect any part of the body, including the eyes and kidneys. Lupus most often affects the heart, joints, skin, lungs, blood vessels, kidneys and central nervous system (CNS). The clinical course is unpredictable and is characterized by periods of remissions and flares, which may be acute or chronic29.
Any structure of the eye can be involved in SLE, with keratoconjunctivitis sicca (KCS) being the most common manifestation as a result of secondary Sjögrens syndrome30. The next most common pathologic condition involving the eye in lupus patients is retinal vasculopathy in the form of cotton wool spots31. Other less common ophthalmologic manifestations of SLE include optic neuropathy, choroidopathy, episcleritis, scleritis, and anterior uveitis (iritis, iridocyclitis)30. Orbital tissues such as the lacrimal gland, extraocular muscles, and other orbital tissues may also be involved in SLE leading to pain, proptosis, lid swelling, and diplopia32. In addition, there are specific ocular toxicities secondary to medications seen in patients with SLE, including glucocorticoid‑induced glaucoma and cataract, and retinal toxicity due to antimalarial therapy29,30.
Renal involvement is clinically apparent in approximately 50 percent of SLE patients, and is a significant cause of morbidity and mortality33. An abnormal urinalysis with or without an elevated plasma creatinine concentration is present in a large proportion of patients at the time of diagnosis of lupus nephritis, but the most frequently observed abnormality in patients with lupus nephritis is proteinuria34. Thus, periodic screening for the presence of lupus nephritis with urinalyses, quantitation of proteinuria, and estimation of the GFR is an important component of the ongoing management of SLE patients33. Several forms of glomerulonephritis can occur, and renal biopsy is useful to define the type and extent of renal involvement. The clinical presentation of lupus nephritis is highly variable ranging from asymptomatic hematuria and/or proteinuria to nephrotic syndrome and rapidly progressive glomerulonephritis with loss of renal function35.
Sjögrens syndrome
Sjögrens syndrome (SS) is an autoimmune disorder caused by the infiltration of monocytes in epithelial glandular and extra‑glandular tissues. Hallmark presentations include mouth and eye dryness, known as KCS. KCS is characterized primarily by a deficiency in tear production, leading to keratinization and a loss of conjunctival goblet cells, resulting in tear mucin deficiency36.
Although renal involvement is uncommon in primary SS, patients may experience several types of kidney injury. Chronic tubulointerstitial nephritis is the most common renal manifestation of SS37. The interstitial nephritis in SS is characterized histologically by an interstitial infiltrate that can invade and damage the tubules38. In some cases, granuloma formation is seen, and there may be a concurrent uveitis, suggesting the possible presence of sarcoidosis or the tubulointerstitial nephritis and uveitis (TINU syndrome)39. The clinical manifestations of the interstitial nephritis include a variable but generally mild elevation in the plasma creatinine concentration, a relatively benign urinalysis, and abnormalities in tubular function, including Fanconi syndrome, distal (type 1) renal tubular acidosis (RTA), nephrogenic diabetes insipidus (tubular resistance to antidiuretic hormone), and hypokalemia40.
Glomerular involvement is much less common than interstitial nephritis in SS. Membranoproliferative glomerulonephritis (MPGN) and membranous nephropathy (MN) are the most common. Other glomerular lesions including minimal change disease, immunoglobulin A (IgA) nephropathy, and antineutrophil cytoplasmic antibody (ANCA)‑associated vasculitis have been reported40.
Behçets syndrome
Behçets syndrome (BS) is a multisystem inflammatory syndrome of unknown etiology characterized by recurrent oral aphthae, genital ulcers, and ocular inflammation. Minor criteria include arthritis, intestinal ulcers, epididymitis, vascular disease, and neuropsychiatric symptoms41.
The disease is most often diagnosed in young adults between the ages of 20 and 40 years, generally in Mediterranean countries, the Middle East, and Japan, but it has been described worldwide. Mostly involved organs and systems are as follows: oral aphthosis (98%), genital aphthosis (73%), skin (74%), eyes (51%), joints (39%), neurological system (7.3%). Other systems, such as the kidneys, are involved less frequently42.
Renal involvement in BS is less frequent and often less severe than in other types of vasculitis. Patients with renal disease may have proteinuria, hematuria, or mild renal insufficiency, but can progress to renal failure.
Although urinary abnormalities (proteinuria and/or hematuria) occur in approximately 10% of patients, serious renal lesions are rare43. The spectrum of renal diseases is wide and can range from AA (secondary) amyloidosis, glomerulonephritis (IgA nephropathy to crescentic glomerulonephritis), vascular disease (mostly renal arterial aneurysms), and interstitial nephritis44.
Ocular disease occurs in 25 to 75% of patients with BS45. Uveitis is often the dominant feature of BS. It is typically bilateral and episodic, often involves the entire uveal tract (panuveitis), and may not resolve completely between episodes. Isolated anterior uveitis is rare.
Hypopyon is seen in about 20% of patients with BS. Many patients with hypopyon will demonstrate retinal vasculitis46.
Posterior uveitis, retinal vasculitis, vascular occlusion, and optic neuritis can also occur and require systemic immunosuppressive treatment and may irreversibly impair vision and progress to blindness if untreated. Other changes that can be seen include neovascularization, secondary cataracts, glaucoma, frosted branch angiitis and conjunctival ulceration47. Conjunctivitis, scleritis, episcleritis, and keratoconjunctivitis sicca are uncommon.
Ocular signs may be helpful in making the diagnosis of BS, even in the absence of other clinical symptoms48.
Vasculitis
Vasculitis are defined by the presence of inflammatory leukocytes in vessel walls with reactive damage to mural structures. Both loss of vessel integrity leading to bleeding, and compromise of the lumen may result in downstream tissue ischemia and necrosis. In general, affected vessels vary in size, type, and location in association with the specific type of vasculitis. Vasculitis may occur as a primary process or may be secondary to another underlying disease49.
A large number of systemic diseases resulting from immunologic pathogenic mechanisms are associated with oculorenal manifestations. The major ocular manifestations of autoimmune disorders are presented in Table I.
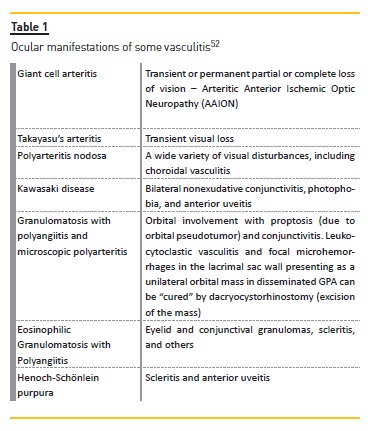
In Hemolytic Uremic Syndrome (HUS) ocular manifestations are rare and include retinal, choroidal, and vitreal hemorrhages, retinal ischemic signs, and non‑perfusion.
Eculizumab was shown to be effective for atypical HUS as it inhibits activation of the terminal complement pathway and seems to be a promising treatment for atypical HUS and its ocular complications50.
Sturm et al. aimed to study the frequency and severity of ocular involvement in pediatric patients with HUS. Three of 69 examined patients with HUS showed ocular involvement, so only a minority of pediatric patients with HUS developed ocular involvement. Acute ocular findings varied in severity from isolated intraretinal hemorrhages to Purtscher‑like retinopathy with retinal ischemia. Long‑term complications included the development of neovascularizations and consecutive optic nerve atrophy. Although ocular involvement in HUS seems to be rare, physicians should be aware of this complication because of its possible vision endangering consequences51.
The kidneys are frequently affected by systemic vasculitis. This is not surprising given the numerous vessels within the renal parenchyma. Usually vasculitis cause renal dysfunction predominantly by inducing glomerular inflammation with resultant nephritis and renal failure49. Detailed clinical manifestations of kidney involvement of all vasculites will not be reviewed in this paper.
Infectious diseases
Tuberculosis
Tuberculosis (TB) is the second most common cause of death from a single infectious agent worldwide. It is a highly contagious, persistent disease characterized by the formation of hard grayish nodules, or tubercles.
The disease is most often caused by the bacterium Mycobacterium tuberculosis and usually occurs in the lungs (the initial site of infection), but it also can occur in other organs, such as the eyes and the kidney53.
In general, TB involving the kidney occurs by mycobacterial seeding of the urogenital tract by hematogenous spread. This occurs at the time of pulmonary infection or in the setting of reactivation or military disease54. Tuberculous bacilli may enter the medullary interstitium and cause granulomas formation. These may heal with associated fibrosis or, many years after initial infection, may break down and rupture into the tubular lumen with excretion of tuberculous bacilli into the urinary tract, leading to continuous spread of infection.
The descending spread of infection to the ureter and bladder causes ureteral stricture and obstruction, hydronephrosis, and renal dysfunction54. Less commonly, interstitial nephritis and glomerulonephritis can occur. The pathogenesis of TB‑associated interstitial nephritis is not clear. It may be an immunologic phenomenon caused by TB or it can be the result of drugs used to treat TB, such as rifampicin. There are also a number of case reports of patients with glomerulonephritis associated with TB. TB can also cause renal amyloidosis, due to chronic inflammation, with high circulating levels of the acute‑phase reactant, serum amyloid A protein55,56.
Initially, renal TB is not associated with specific symptoms. Pyuria and/or microscopic hematuria may be observed as incidental findings. Once the disease has progressed to involve the bladder, symptoms of frequency, dysuria, urgency, and nocturia occur in approximately half of cases. Hematuria and low back pain develop in one‑third of cases57. Systemic symptoms (fever, weight loss) are relatively rare. Manifestations of advanced disease include end‑stage renal disease and, rarely, refractory hypertension58. Characteristic laboratory findings include persistent pyuria and acidic urine in the setting of urine cultures that are repeatedly negative for pyogenic organisms. Painless macroscopic or microscopic hematuria is present in more than 90 percent of cases54. The plasma creatinine concentration is usually normal and an elevated plasma creatinine concentration may be observed in the setting of bilateral renal involvement and/or in the setting of interstitial nephritis or glomerulonephritis57. The diagnosis may be established by demonstration of tubercle bacilli in the urine. Urine mycobacterial culture or urine polymerase chain reaction (PCR) for Mycobacterium tuberculosis should be performed. Radiographic imaging is also warranted for patients with suspected renal TB.
Computerized tomography with contrast is preferred when feasible. Tuberculin skin test or interferon‑gamma release assay and histopathology are also useful in the diagnosis57.
Tuberculosis of the eye may be intraocular or it may involve the external structures. Most commonly, ocular TB develops as a result of hematogenous spread of M. tuberculosis from pulmonary or extrapulmonary sites. The clinical manifestations include choroiditis, chorioretinitis, choroidal granuloma, optic neuritis, optic disc granuloma, subretinal abscess, orbital cellulitis, scleritis, necrotizing scleritis, posterior scleritis, sclerokeratouveitis, interstitial keratitis, and anterior chamber granuloma (59, 60). Less commonly, ocular TB can occur as a result of direct ocular infection from an exogenous source. In such cases, infection may involve the ocular adnexa, lacrimal gland, conjunctiva, sclera, or cornea. In rare cases, eye involvement can occur as a result of a hypersensitivity reaction to a distant focus of infection. Manifestations may include episcleritis, phlyctenulosis, and occlusive retinal vasculitis60.
Genetic disorders
A variety of human congenital syndromes affecting both organs have been described.
Alport syndrome
Alport syndrome is an inherited progressive form of glomerular disease that is often associated with sensorineural hearing loss and ocular abnormalities. It is an X‑linked or autosomal recessive (and extremely rarely autosomal dominant) and a primary basement membrane disorder arising from mutations in genes encoding several members of the type IV collagen protein family61.
Several ocular defects involving the lens, retina, and cornea have been reported in patients with Alport syndrome62. Anterior lenticonus is a regular conical protrusion on the anterior aspect of the lens due to thinning of the lens capsule. It occurs in 20 to 30% of males with X‑linked Alport syndrome and is pathognomonic of the disease. Lenticonus can be complicated by the presence of subcapsular cataracts, which may lead to loss of visual acuity. Retinal changes are usually asymptomatic, and when there is anterior lenticonus, they are always present63. The changes consist of bilateral white or yellow granulations that are superficially located in the retina surrounding the foveal area (also referred to as dot and fleck or fleck retinopathy). These findings are also specific for Alport syndrome. Corneal changes in patients with Alport syndrome can include posterior polymorphous dystrophy and recurrent corneal erosion, which can cause severe ocular pain64,65.
In the kidney, the initial renal manifestation of Alport syndrome is asymptomatic persistent microscopic hematuria, present in early childhood in affected patients. Recurrent episodes of gross hematuria are not uncommon especially during childhood and may be the initial presenting finding and often occurs after an upper respiratory infection66. Boys without hematuria by the age of 10 years are unlikely to have Alport syndrome61. In early childhood, the serum creatinine and blood pressure are normal. Over time, proteinuria, hypertension, and progressive renal insufficiency develop. ESRD usually occurs between the ages of 16 and 35 years in patients with X‑linked or autosomal recessive disease. In some families, the course is more indolent, with renal failure being delayed until age 45 to 60, especially in those with autosomal dominant Alport syndrome. In females with X‑linked Alport syndrome, recurrent episodes of gross hematuria, proteinuria, hearing loss, and diffuse glomerular basement membrane (GBM) thickening are associated with more severe renal dysfunction and
ESRD at an earlier age67.Fabry disease
Fabry disease is a rare X‑linked recessive metabolic defect of alpha‑galactosidase A (α‑Gal A). The absence of α‑Gal A enzyme activity leads to accumulation of glycosphingolipid globotriaosylceramide (GL‑3) in the lysosomes of a variety of cell types. It can cause skin and ocular lesions, progressive renal, cardiac or cerebrovascular disorders68.
Regarding eye disease, the most specific, almost pathognomonic and common finding is cornea verticillata (corneal deposits) but other abnormalities such as vascular tortuosities and posterior subcapsular cataract can occur69.
Renal manifestations occur in at least 50% of male patients and approximately 20% of female patients. The main findings are proteinuria followed by progressive renal insufficiency and often hypertension.
Uncommonly, patients complain of polyuria and polydipsia or they are discovered by the presence of renal sinus cysts on an imaging study68. One of the earlier signs of nephropathy is microalbuminuria.
Over time, proteinuria under the nephrotic range is the most typical form of presentation and is an independent risk factor affecting the extent of renal decline. A proteinuria level above 1 g/day is associated with a worst prognosis69. Other features are glomerular hyperfiltration, impaired concentration ability due to distal tubular involvement, and increased urinary Gb3 excretion. Urinalysis may be quite variable and microscopy may be useful in the diagnosis because vacuolated epithelial cells filled with glycosphingolipids give the appearance of a Maltese cross when polarized light microscopy is used, and they are very specific. Renal ultrasound shows parapelvic cysts in up to 50% of patients. A wide range of renal histopathology could be found due to diffuse deposition of glycosphingolipid in the glomeruli, tubules, and vasculature. Light microscopic findings include a foamy appearance of the glomeruli with diffuse swelling and vacuolization of visceral podocytes, mesangial expansion and progressive segmental and global glomerulosclerosis.
Electron microscopy shows podocytes and mesangial cells filled with lysosomal electron dense granules.
As seen with other nephropathies, glomerular sclerosis and tubulointerstitial fibrosis, although not specific, are the histological features that best correlate with the progression of renal disease. A glomerular sclerosis above 50% predicts a worst prognosis69,70.
Autosomal Dominant Polycystic Kidney Disease
Autosomal dominant polycystic kidney disease (ADPKD) is a common genetic disorder, occurring in approximately 1 in every 400 to 1000 live births. ADPKD is caused by two known (and possibly more unknown) genetic mutations: PKD1 (which encodes polycystin‑1) on chromosome 16 and PKD2 (which encodes polycystin‑2) on chromosome 471.
It has been determined that the embryogenetic stages of eye and kidney development occur rather simultaneously. From the 7th to the 10th week, the development of the ocular architecture progresses in parallel with the differentiation of the kidney tubules72.
It is conceivable that the unknown factor leading to the development of renal tubular cysts in utero might simultaneously affect the eye and the kidney. Meyrier et al.73 reported a strongly association between ADPKD and blepharochalasis. Other ocular abnormalities including severe myopia, cataracts, papilledema, and peripheral retinal pigmentation have also been described in association with ADPKD72.
Virtually all individuals who inherit PKD1 or PKD2 eventually develop renal cysts that are visible by ultrasonographic imaging studies74. The age at which affected individuals have clinical manifestations such as renal failure or hypertension is variable. Patients with PKD1 present with symptoms at a younger age than those with PKD2. Patients with ADPKD can present with hypertension, hematuria, proteinuria, or renal insufficiency, detected by routine laboratory examinations. Flank pain due to renal hemorrhage, calculi, or urinary tract infection is the most common symptom reported by patients.
Hypertension is present in the majority of patients who have normal renal function and have reached the fourth decade of life. Patients may also present with symptoms that are secondary to cysts in other organs, such as the liver, pancreas, spleen, thyroid, or epididymis. The diagnosis is most commonly made in the settings of routine screening in an asymptomatic patient with a positive family history of ADPKD, initial work‑up for new‑onset hypertension, as an accidental finding during an imaging study performed for an unrelated reason and during evaluation of ADPKD‑specific symptoms (hematuria, cyst rupture, pyelonephritis, kidney stones)75.
Nephronopthisis
The most frequent genetic cause of chronic renal failure in children is nephronophthisis. Multiple genes appear to underlie this disorder. In kidney, nephronophthisis is characterized by tubular distention and cysts at the corticomedullary junction tubulointerstitial nephritis, and glomerulosclerosis. Cortical microcysts occur isolated or in combination with chronic tubulointerstitial nephritis in patients with NPHP2 (inversin) gene mutations76. This autosomal recessive disorder is typically associated with severe hypertension and progresses to end‑stage ESRD before two years of age. When associated with retinitis pigmentosa, the renal‑retinal disorder is termed the Senior‑Loken syndrome77.
Other genetic diseases
In addition to the genetic diseases already mentioned involving the kidney and the eye, there are still a number of genetic diseases to mention (Table II).
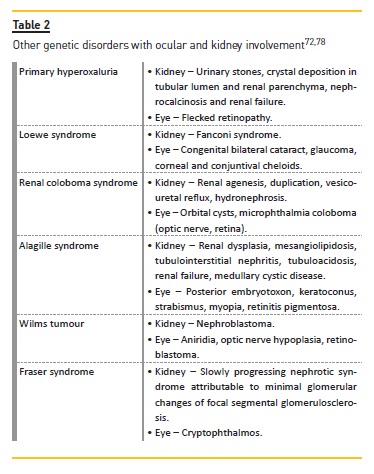
Ocular and renal diseases related to kidneytransplantation
Abnormalities of the eye are common in patients with kidney transplantation. One study found that 42 of 80 renal allograft recipients developed ocular complications including posterior subcapsular cataract, opportunistic ocular infections, raised intraocular pressure79.
The administration of chronic corticosteroids alone may induce multiple eye complications, particularly posterior subcapsular cataracts (79, 80). In a report of 176 renal transplant recipients, a posterior subcapsular cataract was present in 60 (34%)81.
After excluding acute glaucoma, the presence of impaired vision and a painful red eye strongly suggests infection in recipients of solid organ transplants. Nocardial endophthalmitis, cryptococcal choroiditis, cytomegalovirus chorioretinitis, and Listeria monocytogenes endophthalmitis have been reported82. BK virus‑induced or polyomavirus‑associated nephropathy (PVAN) due to the BK virus is an important cause of kidney transplant dysfunction and allograft loss. Treatment with low‑dose intravenous cidofovir may be complicated by anterior uveitis. In 14 kidney‑transplant recipients given low dose cidofovir for PVAN, five (35%) patients developed anterior uveitis. Following discontinuance of cidofovir, there was complete resolution of uveitis in four patients treated with topical corticoids and cycloplegics83. Other medication, such as Tacrolimus can also affect the eye. Preclinical toxicity studies in rats showed that tacrolimus may cause cataract due to an accumulation of sorbitol in the lens secondary to the diabetogenic effect of the drug. Tacrolimus, like cyclosporine, may have a direct neurotoxic effect. Acute cortical blindness has been reported occurring 5‑47 days following transplantation. Reversibility within few weeks after discontinuation of tacrolimus is generally observed. A case of bilateral anterior ischemic optic neuropathy 3 months after liver transplantation has been reported in a patient on tacrolimus therapy. Clinical features resembled ischemic optic neuropathies.
Deterioration of vision and bilateral optic atrophy occurred despite discontinuation of the tacrolimus84.
CONCLUSIONS
In summary, there is a substantial evidence of a close association between kidney and eye diseases and therefore a large number of diseases with both renal and kidney involvement. Some pathologies or signs/symptoms should prompt ophthalmologic referral, such as:
– Stage 4 hypertension (SBP > 210 mmHg or DBP > 120 mmHg);
– Sudden visual loss;
– Painful red eye with loss of vision (especially in patients with autoimmune diseases);
– Diabetic conditions with sudden or severe loss of vision;
On the other hand, there are other situations that need a regular but not urgent ophthalmologic follow‑up:
– Patients with suspected diabetic nephropathy, to evaluate the existence of diabetic retinopathy, since there is a strong correlation between diabetic retinopathy and diabetic nephropathy;
– Patients with lupus nephritis under hydroxychloroquine, to exclude retinal toxicity;
– Patients with Sjogrens Syndrome to assess severity and treat cases of dry eye;
– Patients with chronic corticosteroids use, since they have a higher risk of glaucoma and early cataract.
References
1. Grunwald JE, Alexander J, Maguire M, Whittock R, Parker C, McWilliams K, et al. Prevalence of ocular fundus pathology in patients with chronic kidney disease. Clin J Am Soc Nephrol. 2010;5(5):867–73 [ Links ]
2. Wong CW, Wong TY, Cheng CY, Sabanayagam C. Kidney and eye diseases: common risk factors, etiological mechanisms and pathways. Kidney Int. 2014;85(6):1290–302 [ Links ]
3. Jeng CJ, Hsieh YT, Yang CM, Yang CH, Lin CL, Wang I. Diabetic retinopathy in patients with diabetic nephropathy: development and progression. PLoS ONE 2016;11(8):e0161897 [ Links ]
4. Jawa A, Kcomt J, Fonseca VA. Diabetic nephropathy and retinopathy. Med Clin North Am. 2004;88(4):1001–36 [ Links ]
5. Klein R, Klein BE, Moss SE, Davis MD, DeMets DL. The Wisconsin epidemiologic study of diabetic retinopathy. IV. Diabetic macular edema. Ophthalmology. 1984;91(12):1464–74 [ Links ]
6. Klein R, Klein BE, Moss SE, Davis MD, DeMets DL. The Wisconsin epidemiologic study of diabetic retinopathy. III. Prevalence and risk of diabetic retinopathy when age at diagnosis is 30 or more years. Arch Ophthalmol. 1984;102(4):527–32 [ Links ]
7. Cai X, McGinnis JF. Diabetic retinopathy: animal models, therapies, and perspectives. J Diabetes Res. 2016;2016:3789217 [ Links ]
8. Frank RN. Etiologic mechanisms in diabetic retinopathy. Retina. 1994;Vol 2:1243–76 [ Links ]
9. Ways DK, Sheetz MJ. The role of protein kinase C in the development of the complications of diabetes. Vitam Horm. 2000;60:149–93 [ Links ]
10. Pavenstadt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev. 2003;83(1):253–307 [ Links ]
11. Gruden G, Perin PC, Camussi G. Insight on the pathogenesis of diabetic nephropathy from the study of podocyte and mesangial cell biology. Curr Diabetes Rev. 2005;1(1):27–40 [ Links ]
12. Chawla A, Chawla R, Chawla A. Correlation Between Retinopathy Microalbuminuria and Other Modifiable Risk Factors. Presented on American Diabetes Associations 75th Scientific Session; June 5‑9; Boston, Massachusetts. 2015. [ Links ]
13. Ravera M, Re M, Deferrari L, Vettoretti S, Deferrari G. Importance of Blood Pressure Control in Chronic Kidney Disease. J Am Soc Nephrol. 2006;17:S98–S103 [ Links ]
14. Klag MJ, Whelton PK, Randall BL, Neaton JD, Brancati FL, Ford CE, et al. Blood pressure and end‑stage renal disease in men. N Engl J Med. 1996;334:13–18 [ Links ]
15. Tozawa M, Iseki K, Iseki C, Kinjo K, Ikemiya Y, Takishita S. Blood pressure predicts risk of developing end‑stage renal disease in men and women. Hypertension. 2003;41:1341–5 [ Links ]
16. Bidani AK, Griffin KA. Long‑term renal consequences of hypertension for normal and diseased kidneys. Curr Opin Nephrol Hypertens. 2002;11:73 –80 [ Links ]
17. Keith NM, Wagener HP, Barker NW. Some different types of essential hypertension: their course and prognosis. Am J Med Sci. 1974;268(6):336 [ Links ]
18. Wong TY, Mitchell P. Hypertensive retinopathy. N Engl J Med. 2004;351(22):2310 [ Links ]
19. Baughman RP, Teirstein AS, Judson MA, Rossman MD, Yeager H Jr, Bresnitz EA, et al. Clinical characteristics of patients in a case control study of sarcoidosis. Am J Respir Crit Care Med. 2001;164(10 Pt 1):1885 [ Links ]
20. Evans M, Sharma O, LaBree L, Smith RE, Rao NA. Differences in clinical findings between Caucasians and African Americans with biopsy‑proven sarcoidosis. Ophthalmology. 2007;114(2):325–33 [ Links ]
21. Herbort CP, Rao NA, Mochizuki M, members of Scientific Committee of First International Workshop on Ocular Sarcoidosis. International criteria for the diagnosis of ocular sarcoidosis: results of the first International Workshop On Ocular Sarcoidosis (IWOS). Ocul Immunol Inflamm. 2009; 17(3):160–9 [ Links ]
22. Mavrikakis I, Rootman J. Diverse clinical presentations of orbital sarcoid. Am J Ophthalmol. 2007;144(5):769 [ Links ]
23. Casella FJ, Allon M. The kidney in sarcoidosis. J Am Soc Nephrol. 1993;3(9): 1555 [ Links ]
24. Berliner AR, Haas M, Choi MJ. Sarcoidosis: the nephrologists perspective. Am J Kidney Dis. 2006;48(5): 856–70 [ Links ]
25. Muther RS, McCarron DA, Bennett WM. Renal manifestations of sarcoidosis. Arch Intern Med. 1981;141(5):643 [ Links ]
26. Mackensen F, Billing H. Tubulointerstitial nephritis and uveitis syndrome. Curr Opin Ophthalmol. 2009; 20(6):525–31 [ Links ]
27. Mandeville JT, Levinson RD, Holland GN. The tubulointerstitial nephritis and uveitis syndrome. Surv Ophthalmol. 2001;46(3):195–208 [ Links ]
28. Koike K, Lida S, Usui M, Matsumoto Y, Fukami K, Ueda S et al. Adult‑onset acute tubulointerstitial nephritis and uveitis with Fanconi syndrome. Case report and review of the literature. Clin Nephrol. 2007;67(4):255 [ Links ]
29. Cervera R, Khamashta MA, Font J, Sebastiani GD, Gil A, Lavilla P, et al. Systemic lupus erythematosus: clinical and immunologic patterns of disease expression in a cohort of 1,000 patients. The European Working Party on Systemic Lupus Erythematosus. Medicine (Baltimore) 1993;72:113 [ Links ]
30. Silpa‑archa S, Lee JJ, Foster CS. Ocular manifestations in systemic lupus erythematosus. Br J Ophthalmol. 2016;100(1): 135 [ Links ]
31. Gold DH, Morris DA, Henkind P. Ocular findings in systemic lupus erythematosus. Br J Ophthalmol. 1972;56(11):800 [ Links ]
32. Rosenbaum JT, Trune DR, Barkhuizen A, et al. Ocular, aural, and oral manifestations. In: Dubois lupus erythematosus and related syndromes, Eigth, Wallace DJ and Hahn BH (Ed), Elsevier, Philadelphia 2013:393.
33. Danila MI, Pons‑Estel GJ, Zhang J, Vilá LM, Reveille JD, Alarcón GS. Renal damage is the most important predictor of mortality within the damage index: data from LUMINA LXIV, a multiethnic US cohort. Rheumatology (Oxford). 2009;48(5):542–5 [ Links ]
34. Kelley WN. Clinical features of SLE. In: Textbook of Rheumatology, WB Saunders, Philadelphia 2000 [ Links ]
35. Weening JJ, DAgati VD, Schwartz MM, Seshan SV, Alpers CE, Appel GB, et al. The classification of glomerulonephritis in systemic lupus erythematosus revisited. J Am Soc Nephrol. 2004;15(2):241 [ Links ]
36. Arman F, Shakeri H, Nobakht N, Rastogi A, Kamgar M. A Case of Kidney Involvement in Primary Sjögrens Syndrome. Am J Case Rep. 2017;18:622–6 [ Links ]
37. Kidder D, Rutherford E, Kipgen D, Fleming S, Geddes C, Stewart GA.Kidney biopsy findings in primary Sjögren syndrome. Nephrol Dial Transplant. 2015;30(8):1363–9 Epub [ Links ]
38. Bossini N, Savoldi S, Franceschini F, Mombelloni S, Baronio M, Cavazzana I, et al. Clinical and morphological features of kidney involvement in primary Sjögrens syndrome. Nephrol Dial Transplant. 2001;16(12):2328 [ Links ]
39. Maripuri S, Grande JP, Osborn TG, Fervenza FC, Matteson EL, Donadio JV, et al. Renal involvement in primary Sjögrens syndrome: a clinicopathologic study. Clin J Am Soc Nephrol. 2009;4(9):1423–31 [ Links ]
40. Goules A, Masouridi S, Tzioufas AG, Ioannidis JP, Skopouli FN, Moutsopoulos HM. Clinically significant and biopsy‑documented renal involvement in primary Sjögren syndrome. Medicine (Baltimore). 2000;79(4):241 [ Links ]
41. Zeidan MJ, Saadoun D, Garrido M, Klatzmann D1, Six A, Cacoub P. Behçets disease physiopathology: a contemporary review. Auto Immun Highlights. 2016;7(1):4 [ Links ]
42. Davatchi F. Behçet disease: Global perspective. Indian Journal of Rheumatology. 2007;2:65–71 [ Links ]
43. Altiparmak MR, Tanverdi M, Pamuk ON, Tunç R, Hamuryudan V. Glomerulonephritis in Behçets disease: report of seven cases and review of the literature. Clin Rheumatol. 2002;21(1):14 [ Links ]
44. Akpolat T, Akkoyunlu M, Akpolat I, Dilek M, Odabas AR, Ozen S. Renal Behçets disease: a cumulative analysis. Semin Arthritis Rheum. 2002;31(5):317 [ Links ]
45. Nussenblatt RB. Uveitis in Behçets disease. Int Rev Immunol. 1997;14(1):67 [ Links ]
46. Seyahi E, Melikoglu M, Yazici H. Clinical features and diagnosis of Behcets syndrome. Int J Adv Rheumatol. 2007;5:8. [ Links ]
47. Zamir E, Bodaghi B, Tugal‑Tutkun I, See RF, Charlotte F, Wang RC, et al. Conjunctival ulcers in Behçets disease. Ophthalmology. 2003;110(6):1137 [ Links ]
48. Tugal‑Tutkun I, Onal S, Ozyazgan Y, Soylu M, Akman M. Validity and agreement of uveitis experts in interpretation of ocular photographs for diagnosis of Behçet uveitis. Ocul Immunol Inflamm. 2014;22(6): 461–8 [ Links ]
49. Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum 2013;65:1 [ Links ]
50. R David, S Hochberg‑Klein, R Amer. Resolution of ocular involvement with systemic eculizumab therapy in atypical hemolytic‑uremic syndrome. Eye (Lond). 2013;27(8):997–998 [ Links ]
51. Sturm V, Menke MN, Landau K, Laube GF, Neuhaus TJ. Ocular involvement in paediatric haemolytic uraemic syndrome. Acta Ophthalmol. 2010;88(7):804–7 [ Links ]
52. Izzedine H, Bodaghi B, Launay‑Vacher V, Deray G.Oculorenal manifestations in systemic autoimmune disease. Am J Kidney Dis. 2004;43(2):209–22 [ Links ]
53. Daher EF, Junior G, Barros E. Renal Tuberculosis in the Modern Era. Am J Trop Med Hyg. 2013;88(1):54–64 [ Links ]
54. Simon HB, Weinstein AJ, Pasternak MS, Swartz MN, Kunz LJ. Genitourinary tuberculosis. Clinical features in a general hospital population. Am J Med. 1977;63(3):410 [ Links ]
55. van der Meulen J, de Jong GM, Westenend PJ. Acute interstitial nephritis during rifampicin therapy can be a paradoxical response: a case report. Cases J. 2009;2:6643 [ Links ]
56. Sun L, Yuan Q, Feng J, Yao L, Fan Q, Ma J, et al. Be alert to tuberculosis‑mediated glomerulonephritis: a retrospective study. Eur J Clin Microbiol Infect Dis. 2012;31(5):775–9 [ Links ]
57. Figueiredo AA, Lucon AM, Srougi M. Urogenital Tuberculosis. Microbiol Spectr. 2017 [ Links ]
58. Lima NA, Vasconcelos CC, Filgueira PH, Meissa Kretzmann M, Sindeaux T, NetoI BF, et al. Review of genitourinary tuberculosis with focus on end‑stage renal disease. Rev Inst Med Trop Sao Paulo. 2012;54(1):57–60 [ Links ]
59. Vasconcelos‑Santos DV, Rao PK, Davies JB, Sohn EH, Rao NA. Clinical features of tuberculous serpiginouslike choroiditis in contrast to classic serpiginous choroiditis. Arch Ophthalmol. 2010;128(7):853–8 [ Links ]
60. Gupta A, Bansal R, Gupta V, Sharma A, Bambery P.Ocular signs predictive of tubercular uveitis. Am J Ophthalmol. 2010;149(4):562–70 [ Links ]
61. Kashtan CE. Alport syndrome. An inherited disorder of renal, ocular, and cochlear basement membranes. Medicine (Baltimore). 1999;78(5):338 [ Links ]
62. Shaw EA, Colville D, Wang YY, Zhang KW, Dagher H, Fassett R, et al. Characterization of the peripheral retinopathy in X‑linked and autosomal recessive Alport syndrome. Nephrol Dial Transplant. 2007;22(1):104 [ Links ]
63. Byrne MC, Budisavljevic MN, Fan Z, Self SE, Ploth DW. Renal transplant in patients with Alports syndrome. Am J Kidney Dis. 2002;39(4):769 [ Links ]
64. Perrin D, Jungers P, Grünfeld JP, Delons S, Noël LH, Zenatti C. Perimacular changes in Alports syndrome. Clin Nephrol. 1980;13(4):163 [ Links ]
65. Shah SN, Weinberg DV. Giant macular hole in Alport syndrome. Ophthalmic Genet. 2010;31(2):94–7 [ Links ]
66. Gubler M, Levy M, Broyer M, Naizot C, Gonzales G, Perrin D, et al. Alports syndrome. A report of 58 cases and a review of the literature. Am J Med. 1981;70(3):493 [ Links ]
67. Gubler MC. Inherited diseases of the glomerular basement membrane. Nat Clin Pract Nephrol. 2008;4(1):24 [ Links ]
68. Germain DP. Fabry disease. Orphanet J Rare Dis. 2010;5:3 [ Links ]
69. Guedes M, Mira F, Ferreira F, Pinto H, Maia P, Mendes T et al. Fabrys disease, an eye‑kidney disease review. Port J Nephrol Hypert. 2015;29(1):15–20 [ Links ]
70. Branton MH, Schiffmann R, Sabnis SG, Murray GJ, Quirk JM, Altarescu G, et al. Natural history of Fabry renal disease: influence of alpha‑galactosidase A activity and genetic mutations on clinical course. Medicine (Baltimore). 2002;81(2):122 [ Links ]
71. Davies F, Coles GA, Harper PS, Williams AJ, Evans C, Cochlin D. Polycystic kidney disease re‑evaluated: a population‑based study. Q J Med. 1991;79(290):477 [ Links ]
72. Izzedine H, Bodaghi B, Launay‑vacher V, Deray G. Eye and Kidney: From Clinical Findings to Genetic Explanations. J Am Soc Nephrol. 2003;14:516–29 [ Links ]
73. Meyrier A, Simon P. Drooping upper eyelids and polycystic kidney disease. J Am Soc Nephrol. 1994;5:1266–70 [ Links ]
74. Igarashi P, Somlo S. Genetics and pathogenesis of polycystic kidney disease. J Am Soc Nephrol. 2002;13(9):2384 [ Links ]
75. Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet. 2007;369(9569):1287 [ Links ]
76. Gagnadoux MF, Bacri JL, Broyer M, Habib R. Infantile chronic tubulo‑interstitial Nephritis with cortical microcysts: variant of nephronophthisis or new disease entity? Pediatr Nephrol. 1989;3(1):50 [ Links ]
77. Chaki M, Hoefele J, Allen SJ, Ramaswami G, Janssen S, Bergmann C, et al. Genotype‑phenotype correlation in 440 patients with NPHP‑related ciliopathies. Kidney Int. 2011;80(11):1239–45 [ Links ]
78. Hoppe B. An update on primary hyperoxaluria. Nat Rev Nephrol. 2012;8(8):467–75 [ Links ]
79. Das T, Gupta A, Sakhuja V, Gupta KL, Minz M, Chugh KS. Ocular complications in renal allograft recipients. Nephrol Dial Transplant. 1991;6:649 [ Links ]
80. Veenstra DL, Best JH, Hornberger J, Sullivan SD, Hricik DE. Incidence and long‑term cost of steroid‑related side effects after renal transplantation. Am J Kidney Dis. 1999;33:829 [ Links ]
81. Matsunami C, Hilton AF, Dyer JA, Rumbach OW, Hardie IR. Ocular complications in renal transplant patients. Aust N Z J Ophthalmol. 1994;22:53 [ Links ]
82. Suppiah R, Abraham G, Sekhar U, Mathew M, Shroff S, Soundararajan P. Nocardial endophthalmitis leading to blindness in a renal transplant recipient. Nephrol Dial Transplant 1999;14:1576 [ Links ]
83. Lopez V, Sola E, Gutierrez C, Burgos D, Cabello M, García I, et al. Anterior uveitis associated with treatment with intravenous cidofovir in kidney transplant patients with BK virus nephropathy. Transplant Proc 2006;38:2412 [ Links ]
84. Lanzetta P, Monaco P. Major ocular complications after organ transplantation. Transplant Proc. 2004;36(10):3046‑8 [ Links ]
Rachele Escoli, MD
Centro Hospitalar do Médio Tejo
Avenida Xanana Gusmão, Ap 45, 2350‑754,
Torres Novas
E‑mail: rachele_escoli@hotmail.com
Disclosure of potential conflicts of interest: none declared
Received for publication: Feb 19, 2018
Accepted in revised form: Jul 15, 2018

