










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
versão impressa ISSN 0872-0169
Port J Nephrol Hypert vol.32 no.4 Lisboa dez. 2018
REVIEW ARTICLE
Congenital kidney and urinary tract anomalies: a review for nephrologists
Marina Vieira, Aníbal Ferreira, Fernando Nolasco
Nephrology Department, Hospital Curry Cabral, Centro Hospitalar Lisboa Central, Lisboa, Portugal
ABSTRACT
Kidney and urinary tract development disorder are two of the most prevalent congenital malformations and the main cause of chronic kidney disease in pediatric age patients. As such, it is very important that the nephrologist understands these pathologies to improve transition and ensure a good continuity between pediatric and adult nephrological care.
The purpose of this article is to present a brief review of congenital anomalies of the kidney and urinary tract (CAKUT).
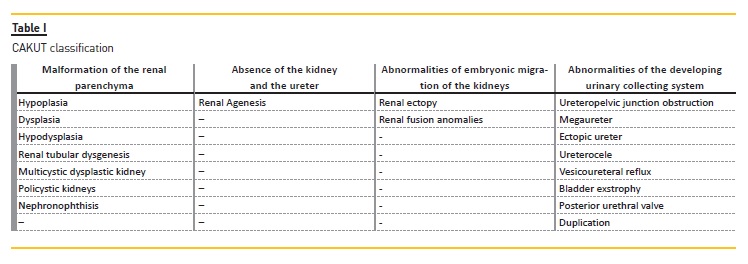
Kidney malformations are classified according to macroscopic and microscopic anatomic features, and are the result of the following abnormal renal developmental processes: malformations of the renal parenchyma, abnormalities of the embryonic migration of the kidneys and abnormalities of the developing urinary collecting system.
Keys words: congenital anomalies of the kidneys and urinary tract, dysplasia, ciliopathies, posterior urethral valves, vesicoureteral reflux.
INTRODUCTION
Kidney and urinary tract development disorders include a spectrum of malformations ranging from the complete absence of renal tissue to minor structural alterations1. Structural developmental anomalies are the most frequently diagnosed diseases in the prenatal period, accounting for 20 to 30% of cases2,3 and account for 30‑50% of all cases of end‑stage kidney disease (ESKD) in children4. In adults, CAKUTs account for only 2‑3% of ESKD and these patients need to initiate a renal replacement technique much earlier (mean age 30 years vs. 61 years) compared to the other causes of ESKD5.
The risk of ESKD differs depending on the causes of CAKUT. Patients with a functioning single kidney or renal hypodysplasia associated with posterior urethral valves are more likely to require dialysis6. Kidney malformations are classified according to macroscopic and microscopic anatomic features, and are the result of the following abnormal renal developmental processes:
– Malformation of the renal parenchyma
– Absence of the kidney and the ureter (which can be included in renal parenchyma abnormalities)
– Abnormalities of embryonic migration of the kidneys
– Abnormalities of the developing urinary collecting system
Boys are 1.3‑1.9 times more likely to be affected than girls7. CAKUT are associated with malformation syndromes in about 30 percent of cases8, and there are more than 200 syndromes which include CAKUT and nonrenal anomalies9.Table I
MALFORMATION OF THE RENAL PARENCHYMA
Normal renal development is dependent upon the interaction between the ureteric bud and metanephric mesenchyme, which induces organogenesis. The pathogenesis of renal parenchymal malformations is thought to be multifactorial and involve genetic as well as environmental factors10‑12.
Renal hypoplasia is defined as renal length less than two standard deviation below the mean for age associated with reduction in number of nephrons but a normal renal architecture. In renal hypoplasia the kidney has only 20 to 25 percent of the normal total number of nephrons13 with hypertrophic glomeruli and tubules, thickening of Bowmans capsule and variable abnormalities of the glomerular basement membrane (fusion of epithelial cell foot processes)14. If bilateral and severe, this can lead to ESKD.
Renal dysplasia are malformations of different renal tissue, with or without cysts. Most dysplastic kidneys are small for the patient´s age, and vary as to the presence of epithelial cysts (number and size). Cystic dysplasia is often associated with antenatal obstruction of the urinary tract, which may be associated with posterior urethral valves (PUV) or ureteropelvic junction obstruction (UPJO)15.
Multicystic dysplastic kidney (MCDK) is a severe form of renal dysplasia in which very large cysts (noncommunicating) dominate kidney structure and normal renal tissue cannot be identified. The cause of MCDK is unclear and a genetic reason may underlie it. A study that analyzed coding exons of genes associated with CAKUT in a large cohort of children reported MCDK was associated with mutations in the proto‑oncogenes (CHD 1L, ROBO2, HNF 1B, SALL1 and PAX2)16. MCDK may involve both kidneys, but most cases are unilateral, and in the majority of cases, an involution of the disease occurs as demonstrated by repeat ultrasound exam17. If the contralateral kidney is normal, it frequently undergoes compensatory hypertrophy, which begins early in utero and results in a kidney size that is greater than two standard deviations beyond the mean length. The absence of compensatory hypertrophy suggests abnormalities. Because most cases of MCDK involute, conservative management with long‑term ultrasound echographic follow‑up is recommended, as individual patients with contralateral abnormalities may develop renal impairment.
The term hypodysplasia is utilized in cases of congenitally small kidney (reduced number of nephrons) with dysplastic features. Renal hypoplasia is more commonly associated with dysplasia than without.
Renal tubular dysgenesis (RTD) may be acquired during fetal development or inherited as an autosomal recessive disease. It is a special form of dysplasia with anomalous renal tubular development (absence or paucity of differentiated proximal tubules) secondary to the reduction of perfusion of the embryonic kidney and accompanied by thickening of renal arterial vasculature.
Genetically RTD results from mutations in the genes encoding the major components of the renin‑angiotensin system: angiotensinogen, renin, angiotensin‑converting enzyme or angiotensin II receptor type 1. Consequently there is absence of production or lack of efficacy of angiotensin II18. Prenatal clinical manifestations are persistent anuria with oligohydramnios and pulmonary hypoplasia. It is also associated with skull ossification defects (Potter syndrome). At birth, blood pressure is dramatically low and perinatal death occurs in most cases19.
Renal agenesis is the result of absent kidney because it never developed or because of complete involution of a dysplastic kidneys. The correct term is aplasia, for example in MCDK. It may be unilateral or bilateral.
Severe bilateral renal agenesis is incompatible with extrauterine life because prolonged absence of amniotic fluid results in pulmonary hypoplasia, leading to severe respiratory insufficiency at birth.
Renal cysts are present in a many different renal diseases in children, and the term polycystic kidney disease describe the following hereditary conditions:
– Autosomal recessive polycystic kidney disease (ARPKD), previously named infantile polycystic kidney disease, characterized by cystic dilations of the renal collecting ducts and congenital hepatic fibrosis.
– Autosomal dominant polycystic kidney disease (ADPKD), previously named adult polycystic kidney disease, characterized by cystic dilations in all parts of the nephron. Cysts in the liver and pancreas are also common, with most patients with ADPKD presenting when adults; however, some patients present in early childhood and rarely as early as in utero.
The incidence of ADPKD is greater than ARPKD, occurring in one in every 400 to 1000 live births. ADPKD affects up to 12 million individuals and is the 4th most common reason for renal replacement therapy worldwide20.
The disease results from a mutation in the PKD1 gene, located on chromosome 16 which encodes polycystin1, and is present in 85 percent of patients with ADPKD. Other patients with ADPKD have a mutation in the PKD2 gene which encodes polycystin 2 and is located on chromosome 4. Both proteins are involved with cell calcium signaling and localize to the primary cilia of renal epithelial cells (called ciliopathies). ADPKD is characterized by slow development and growth of cysts causing progressive kidney enlargement, with hypertension, abdominal pain, episodes of cyst hemorrhage, nephrolithiasis, cyst infections, extrarenal manifestations and potentially serious complications such as massive hepatomegaly and intracranial aneurysm rupture21. Because of the compensatory hyperfiltration in surviving glomeruli, renal function maintains within the normal range for decades. Renal function declines only when the majority of nephrons have been destroyed, typically after the fourth decade of life, and ESRD eventually ensues22. Most pediatric patients with ADPKD are asymptomatic. Those who do present in childhood have similar renal findings to adult patients.
In contrast, the extrarenal manifestations frequently seen in adults (cysts in the liver and pancreas) are rarely observed in pediatric patients.
ARPKD is always associated with biliary dysgenesis due to an anomalous development which results in persistence of embryologic bile duct structures that become massively dilated. This leads to varying degrees of dilatation of the intrahepatic bile ducts (Caroli disease) and hepatic fibrosis23. Clinical presentation of ARPKD varies based on the onset age of symptoms and the predominance of hepatic or renal involvement24,25.
A moderate genotype‑phenotype correlation is observed; severe phenotypes (perinatal death) are associated with truncating mutations and amino acid substitutions are associated with a nonlethal presentation.
The diagnosis of ADPKD and ARPKD is usually based on family history, clinical and imaging findings and if necessary molecular genetic analysis. Treatment of ARPKD is limited to supportive therapy (management of fluids and electrolytic substitution, control of blood pressure, prevention of chronic kidney disease and renal replacement therapy if necessary)25. Treatment of ADPKD is based on controlling blood pressure and lowering lipid levels in young patients angiotensin‑converting enzyme inhibition (ACEI) and use of tolvaptan vasopressin type 2 receptor antagonist to preserve renal function. Tolvaptan has demonstrated significant reduction of cyst growth and better preservation of glomerular filtration rate within 2 years of treatment period26. In pediatric patients, pravastatin administered in combination with ACEI significantly attenuates the increase in total kidney volume within a 3‑year treatment27.
Ciliopathy is the term used for diseases of the primary cilia, which includes polycystic kidney diseases, medullary cystic kidney disease, and nephronophthisis‑related ciliopathies28. This term describes a group of rare autosomal recessive cystic kidney diseases typically characterized by chronic kidney disease progressing to ESKD during childhood and includes isolated nephronophthisis (NPHP) and certain syndromes with additional extrarenal manifestations, including Senior‑Loken syndrome, Joubert syndrome, Jeunes syndrome, and Meckel‑Gruber syndrome. More than 80 gene‑encoding proteins involved in ciliary structure and function have been associated with the different forms of nephronophthisis‑related ciliopathies29.
Nephronophthisis (NPHP) is the most frequent genetic cause of ESKD in children, and is an autosomal recessive disorder. The first clinical manifestation is an impaired urine‑concentrating ability which causes polyuria, polydipsia and sodium reabsorption. Urinalysis does not have changes and patients develop chronic tubulointerstitial nephritis with posterior progressive ESKD occurring in the first 2 decades of life. The disease variants are infantile, juvenile, and adolescent form with ESKD reached at different median age (1, 13, and 19 years, respectively)30. These different forms have different phenotypic variants which reflect mutations in distinct genes encoding for the nephrocystins, a family of proteins involved in primary ciliary function. The nephrocystins interact with other proteins (e.g., tensin, filamins, tubulins) involved in cell‑cell and cell‑matrix signaling. Mutations in the NPHP genes alter ciliary function (via abnormalities in intracellular signaling pathways) result in the incapacity of the ciliary mechanosensors to sense and regulate luminal flow rates, dysregulation of tissue growth, and cyst formation correctly28,30.
Ultrasound imaging demonstrates increased echogenicity with loss of corticomedullary differentiation.Extrarenal manifestations reflect multisystem disease due to ciliary dysfunction and occur in approximately 20 percent of patients with NPHP31. These manifestations include skeletal defects (scoliosis, polydactyly, brachydactyly and shortening of the limbs and ribs); eye abnormalities (retinitis pigmentosa with Senior‑Loken syndrome); neurologic anomalies (cerebral ataxia, hypotonia, and severe developmental delay); liver involvement (hepatosplenomegaly and portal fibrosis) and cardiac defects (situs inversus and valve abnormalities). The diagnosis of NPHP is suggested by characteristic clinical findings and confirmed by a positive genetic test31. In the early stage of the disease, management of children without renal impairment is supportive and focused on maintaining fluid and electrolyte balance, treating anemia, and promoting normal growth. When glomerular filtration rate decreases, the treatment is similar to that of chronic renal disease, with administration of erythropoietin and iron to patients if anemia is present, providing active vitamin D analogues supplementation and phosphate binder to patients if necessary, and other medication in case of secondary hyperparathyroidism.
Patients with NPHP uniformly progress to ESRD. Renal transplantation is almost the preferred replacement therapy because outcome is excellent, and the disease does not recur in the transplanted kidney32.
ABNORMALITIES OF EMBRYONIC MIGRATION OF THE KIDNEYS
Ectopic kidney encompasses abnormalities in migration. During embryogenesis, the developing kidneys migrate from the pelvis to the retroperitoneal renal fossa and rotate from a horizontal to vertical position.
Disruption of the normal embryologic migration of the kidneys results in the pelvic kidney phenotype (fails to ascend)33. Children with ectopia kidney are generally asymptomatic, and the diagnosis is often made coincidentally during routine antenatal or postnatal ultrasonography. In few cases, children can present symptoms related with complications, such as infection, renal calculi, and urinary obstruction.
Ectopic kidneys are commonly associated with urinary tract abnormalities, particularly with vesicoureteral reflux (VUR) which is present in 30 percent of simple and in 20 percent of crossed ectopic kidneys34.
Renal fusion anomalies are believed to arise between weeks 4 and 9 of gestation, before the kidneys ascend from the pelvis to the dorsolumbar space. The most common configuration is the horseshoe kidney, in which fusion usually occurs at the lower poles of each kidney34. The majority of patients with horseshoe kidneys are asymptomatic. In these patients, the horseshoe kidney is diagnosed by coincidence (e.g., routine antenatal ultrasonography). In antenatal ultrasonography, the most likely cause of an absent kidney in the renal fossa is an ectopic kidney35. Approximately 80 percent of children with horseshoe kidneys have hydronephrosis36.
Causes of hydronephrosis include VUR, obstruction of the collecting system by ureteropelvic junction obstruction (UPJO) or renal calculi37. In the majority of cases, patients have an excellent prognosis without any therapeutic intervention. In patients with VUR associated (grade III‑V) to horseshoe kidneys, prophylactic antibiotic therapy to prevent recurrence of urinary tract infection should be considered. Antimicrobial prophylaxis was associated with a substantially reduced risk of recurrence but not of renal scarring38.
Antimicrobial agents most commonly used for prophylaxis include TMP‑SMZ (2 mg/kg), or nitrofurantoin (1‑2 mg/kg) 48. One daily dose is administered at bedtime.
ABNORMALITIES OF THE DEVELOPING URINARY COLLECTING SYSTEM
Fetal hydronephrosis is the most common anomaly detected on antenatal ultrasonographic examination, affecting 1% to 5% of pregnancies39. UPJO is believed to account for 35% to 50% of these uropathies. The narrowing of the ureteropelvic portion of the ureter may be intrinsic (i.e., caused by fibrosis or by smooth muscle hypertrophy, and abnormal innervation) or extrinsic (i.e., related to aberrant crossing of renal pole vessels or fibrosis of adjacent tissue). Consequently, the obstruction of urine flow leads to dilation of the renal pelvis. The spectrum of phenotypes ranges from slight pelvic dilation with normal urine flow to an almost complete obstruction with renal parenchymal damage and atrophy40. Patients with UPJO are considered at risk of pressure‑induced renal damage. However, congenital obstruction resolves spontaneously in most cases. Nevertheless, individual cases of late deterioration of kidney function have been reported41. A mild pelvic dilation on ultrasonography (<15 mm), more than 40% partial function and, more than 50% nuclide drainage on diuresis renography are considered indicators of a low risk of renal damage. Most children are monitored using ultrasonographic studies and nuclide scans in case of ultrasonographic deterioration at 3‑to 6‑month intervals. When needed, laparoscopic pyeloplasty is preferred because it is minimally invasive, safe, and effective42.
Megaureter can be classified into primary megaureter when the result of a functional or anatomical abnormality involving the ureterovesical junction and into secondary megaureter when it results from abnormalities that involve the bladder or urethra (e.g., myelomeningocele/neurogenic bladder, and posterior urethral valves). In children, any ureter greater than 7 mm in diameter is considered a megaureter based on measurements in fetuses (greater than 30 weeks gestation) and children (<12 years)43. Primary megaureter is the second most common cause of hydronephrosis in newborns (after ureteropelvic obstruction), accounting for approximately 20 percent of cases44. The diagnosis of primary megaureter is made by prenatal or neonatal ultrasonography. Postnatal presentation can occur at any age, with symptoms of UTI, hematuria and abdominal pain. The prognosis of primary megaureter is generally good. Most cases resolve spontaneously within the first 3 years of life. In children with high‑grade hydronephrosis, the condition may persist and may require surgical reinsertion of the ureter45.
Posterior urethral valves (PUV) are obstructing membranous folds within the lumen of the posterior urethra or, in other words, tissue leaflets fanning distally from the prostatic urethra to the external urinary sphincter. PUV are the most common cause of neonatal lower urinary tract obstruction in males, occurring in 1 in 5000 to 8000 pregnancies46. The cause is not completely
understood but it appears that the normal embryologic development of the male urethra is disrupted (between weeks 9 and 14 of gestation) which results in persistence of the urogenital membrane. Posterior urethral valves act as rigid bands or membranes causing obstruction and proximal dilation47.
Prenatal ultrasonography is diagnostic. Renal and urologic manifestations are common in patients with PUV.
They include chronic kidney disease, vesicoureteral reflux (VUR), and bladder dysfunction. The urethral resistance causes detrusor hypertrophy with bladder enlargement and trabeculation, and several formations of pseudodiverticula. It then results in secondary unilateral or bilateral VUR with hydronephrosis, and, at the end disease stage, a renal dysplasia develops. About 1/3 to 1/2 of patients with PUV presents VUR48. VUR is due to the increased intravesical pressure from bladder outlet obstruction, and is classified as secondary cause. Prenatal surgery (bladder marsupialization, valve ablation, vesicoamniotic shunt placement) to relieve obstructive uropathy has been explored but has a morbidity rate and does not improve renal outcomes49.
Nevertheless, primary valve ablation is possible at neonatal age50. Vesicostomy is the next preferred procedure, if valve ablation cannot be performed within the first few months of life. But abnormal bladder function often persists, requiring intermittent catheterization and anticholinergic medication51. Renal damage occurs due to consequent renal dysplasia and possibly due to infection and bladder dysfunction50. One third of patients develop significant CKD and 15% to 20% eventually progress to ESKD52.
Vesicoureteral reflux (VUR) is the retrograde passage of urine from the bladder into the upper urinary tract.
It is divided into two categories: primary and secondary, based on the underlying pathogenesis. The most common form of reflux is primary reflux and is due to incompetence of the ureterovesical junction (UVJ). This UVJ contains a segment of the ureter within the bladder wall (intravesical ureter) which normally prevents reflux during bladder contraction by fully compressing and sealing it off with the surrounding bladder muscles. In primary VUR, failure of this anti‑reflux mechanism is due to a congenitally short intravesical ureter (possibly genetically determined).
Primary VUR is the most common CAKUT entity, with an estimated prevalence of 1% to 6%. The prevalence increases for neonates with prenatal hydronephrosis (up to 15 percent)53,54 and for children with febrile urinary tract infections (UTIs) (ranging from 30 to 45 percent)55. Secondary VUR is a consequence of abnormally high voiding pressure in the bladder that results in failure of the closure of the UVJ during bladder contraction.
Secondary VUR is often associated with anatomic (e.g. posterior urethral valves) or functional
bladder obstruction (bladder bowel dysfunction and neurogenic bladder)56. The degree and chronicity of obstruction can influence the severity of VUR. There is a possible genetic predisposition for primary VUR, although the genetic loci and the mode of inheritance are unknown. It seems that the length of the intravesical ureter may be genetically dictated, but genome linkage has failed to detect any association with known genes57. Ultrasonography is a useful initial investigation to evaluate kidney size, presence of hydroureteronephrosis, bladder wall thickness, and extent of bladder emptying. Staging systems that allow quantification of the severity of reflux (grades I through V) have been defined. The voiding cystourethrogram (VCUG) is the test of choice to establish the presence and degree of VUR. The International Reflux Study Group (IRSG) developed a classification system that grades the severity of VUR based upon the degree of retrograde filling and dilation of the renal collecting system demonstrated by VCUG, but there is an important subjectivity of evaluating VUR grades because there is no perfect concordance (among expert readers) when differentiating between intermediate grades (II and III)58. The different grades of VUR are classified as grade I (reflux only fills the ureter without dilation), grade II (reflux fills the ureter and the collecting system without dilation), grade III (reflux fills and mildly dilates the ureter and the collecting system with mild blunting of the calyces), grade IV (reflux fills and grossly dilates the ureter and the collecting system with blunting of the calyces; some tortuosity of the ureter is also present), grade V (massive reflux grossly dilates the collecting system, all the calyces are blunted with a loss of papillary impression, and intrarenal reflux may be present). VUR grading has a prognostic relevance: spontaneous resolution is observed during the first decade of life in 100% of cases with unilateral and in 50% of those with bilateral grade III VUR and in 40% of those with bilateral grade IV VUR59. There is spontaneous VUR resolution in the majority of cases with primary VUR60.
Children are at risk for recurrent febrile or symptomatic urinary tract infections (UTIs), especially those with more severe VUR. The Randomized Intervention for Vesicoureteral Reflux (RIVUR) trial showed that antibiotic prophylaxis lowered the risk of recurrent symptomatic UTIs. Recently a reanalysis of RIVUR data found that patients who were classified as high‑risk for recurrent UTI (uncircumcised males, those with grade IV VUR, or any child with bladder and bowel dysfunction) derived the most benefit38. These results support a selective approach for identifying children who will most benefit from antibiotic prophylaxis and avoiding unnecessary therapy for others.
CONCLUSION
The kidney and the urinary system are frequent locations of congenital anomalies of differing severity. Many abnormalities are asymptomatic and are diagnosed by prenatal ultrasound or during a systematic evaluation for other congenital anomalies. Sometime these abnormalities are diagnosed as secondary to obstruction, infection or trauma. Boys are more likely to be affected than girls.
Malformations of the renal parenchyma result in abnormality in renal development with the multicystic dysplastic kidney the most severe form of renal dysplasia.
Nephronophthisis is the most frequent genetic cause of ESKD in children and the adolescent variant form of the disease presents at an average of 19 years.
Primary vesicoureteral reflux is the most common CAKUT entity and is due to incompetence of the ureterovesical junction. There is spontaneous VUR resolution in the majority of cases, but selected children are at risk for recurrent febrile urinary tract infections and need antibiotic prophylaxis.
Because CAKUT is the main cause of chronic renal disease in children, and these patients are transferred to adult units, nephrologists must be aware of these pathologies. This knowledge allows a continuity of nephrological follow‑up from paediatric to adult care services. It is also essential to maintain a close and continuous collaboration between adult and pediatric units.
Referências
1. Rodriguez MM, Congenital Anomalies of the Kidney and the Urinary Tract (CAKUT), Fetal Pediatr Pathol 2014;124:293‑320. [ Links ]
2. Loane M, Dolk H, Kelly A, Teljeur C, Greenlees R, Densem J, and a EUROCAT Working Group Paper 4: EUROCAT Statistical Monitoring: Identification and Investigation of Ten Year Trends of Congenital Anomalies in Europe. Birth Defects Research (Part A) 2011;91:31‑43. [ Links ]
3. Queisser‑Luft A, Stolz G, Wiesel A, Schlaefer K, Spranger J, Malformations in newborn: results based on 30,940 infants and fetuses from the Mainz congenital birth defect monitoring system (1990‑1998). Arch Gynecol Obstet 2002;266:163‑167. [ Links ]
4. Seikaly MG, Ho PL, Emmett L, Fine RN, Tejani A, Chronic renal insufficiency in children: the 2001 Annual Report of the NAPRTCS, Pediatr Nephrol 2003;18:796‑804. [ Links ]
5. Wuhl E, Van Stralen KJ, Verrina E, et al. Timing and outcome of renal replacement therapy in patients with congenital malformations of the kidney and urinary tract, Clin J Am Soc Nephrol 2013;8:67‑74. [ Links ]
6. Sanna‑Cherchi S, Pietro Ravani P, Corbani V, et al. Renal outcome in patients with congenital anomalies of the kidney and urinary tract. Kidney International 2009;76:528–533. [ Links ]
7. Harris J, Robert E, Källén B, Epidemiologic characteristics of kidney malformations. Eur J Epidemiol. 2000;16(11):985‑92. [ Links ]
8. Wiesel A, Queisser‑Luft A, Clementi M, Bianca S, Stoll C, EUROSCAN Study Group, Prenatal detection of congenital renal malformations by fetal ultrasonographic examination: an analysis of 709,030 births in 12 European countries. Eur J Med Genet 2005;48:131‑144. [ Links ]
9. Limwongse C, Cassidy SB, Syndromes and malformations of the urinary tract. In: Pediatric Nephrology, 5th ed, Avner ED, Harmon WE, Niaudet P (Eds), Williams & Wilkins, Philadelphia 2004:93‑108. [ Links ]
10. Sanna‑Cherchi S, Caridi G, Weng PL, et al, Genetic approaches to human renal agenesis/hypoplasia and dysplasia. Pediatr Nephrol. 2007;22:1675‑1684. [ Links ]
11. Piscione TD, Rosenblum ND, The malformed kidney: disruption of glomerular and tubular development. Clin Genet 1999;56:341‑356. [ Links ]
12. Martinovic J, Benachi A, Laurent N, Daikha‑Dahmane F, Gubler MC, Fetal toxic effects and angiotensin‑II‑receptor antagonists. Lancet 2001;358:241. [ Links ]
13. Royer P, Habib R, Mathieu H, Courtecuisse V, Congenital bilateral renal hyperplasia with reduction of the number and hypertrophy of the nephrons in children. Ann Pediatr (Paris) 1962;9:133‑139. [ Links ]
14. Morita T, Wenzl J, McCoy J, Porch J, Kimmelstiel P, Bilateral renal hypoplasia with oligomeganephronia: quantitative and electron microsopic study. Am J Clin Pathol 1973;59:104‑112. [ Links ]
15. Chen RY, Chang H, Renal dysplasia. Arch Pathol Lab Med 2015;139:547‑551. [ Links ]
16. Hwang DY, Dworschak GC, Kohl S, et al. Mutations in 12 known dominant disease‑causing genes clarify many congenital anomalies of the kidney and urinary tract. Kidney Int 2014;85:1429‑1433. [ Links ]
17. Hayes WN, Watson AR, Unilateral multicystic dysplastic kidney: does initial size matter? Pediatr Nephrol 2012;27:1335‑1340. [ Links ]
18. Gubler MC, Renal tubular dysgenesis. Pediatr Nephrol 2014;29:51‑59. [ Links ]
19. Gubler MC, Antignac C, Renin‑angiotensin system in kidney development: renal tubular dysgenesis. Kidney Int. 2010;77:400‑406. [ Links ]
20. Chapman AB, Devuyst O, Eckardt KU, et al. Autosomal Dominant Polycystic Kidney Disease: Executive Summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int 2015; 88:17‑27. [ Links ]
21. Torres VE, Harris PC, Pirson Y, Autosomal dominant polycystic kidney disease. Lancet 2007;369:1287‑1301. [ Links ]
22. Grantham JJ, Chapman AB, Torres VE, Volume progression in autosomal dominant polycystic kidney disease: the major factor determining clinical outcomes. Clin J Am Soc Nephrol 2006;1:148‑157. [ Links ]
23. Kamath BM, Piccoli DA, Heritable disorders of the bile ducts. Gastroenterol Clin North Am 2003;32:857‑875. [ Links ]
24. Gunay‑Aygun M, Font‑Montgomery E, Lukose, et al. Correlation of kidney function, volume and imaging findings, and PKHD1 mutations in 73 patients with autosomal recessive polycystic kidney disease. Clin J Am Soc Nephrol 2010;5:972‑984.
25. Guay‑Woodford LM, Bissler JJ, Braun MC, et al. Consensus expert recommendations for the diagnosis and management of autosomal recessive polycystic kidney disease: report of an international conference. J Pediatr 2014;165:611‑617. [ Links ]
26. Torres VE, Chapman AB, Devuyst O, et al. TEMPO 3:4 Trial Investigators: tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med 367:2407‑2418. [ Links ]
27. Cadnapaphornchai MA, George DM, McFann K, et al. Effect of pravastatin on total kidney volume, left ventricular mass index, and microalbuminuria in pediatric autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol 2014;9:889‑896. [ Links ]
28. Hildebrandt F, Benzing T, Katsanis N, Ciliopathies. N Engl J Med. 2011;364:1533‑1543. [ Links ]
29. Devuyst O, Knoers NV, Remuzzi G, et al. Board of the Working Group for Inherited Kidney Diseases of the European Renal Association and European Dialysis and Transplant Association: Rare inherited kidney diseases: challenges, opportunities, and perspectives. Lancet. 2014;383:1844‑1859. [ Links ]
30. F Hildebrandt, M Attanasio, E Otto, Nephronophthisis: disease mechanisms of a ciliopathy. J Am Soc Nephrol 2009;20:23‑35.
31. Srivastava S, Sayer JA, Nephronophthisis. J Pediatr Genet 2014;3:103‑114. [ Links ]
32. Stavrou C, Deltas CC, Christophides TC, Pierides A, Outcome of kidney transplantation in autosomal dominant medullary cystic kidney disease type 1. Nephrol Dial Transplant 2003;18:2165‑2169. [ Links ]
33. Shorecki K, Chertow GM, Marsden PA, Taal MW, Yu ASL Brenner and Rector´s The Kidney 2016; 2311‑2316. [ Links ]
34. Guarino N, Tadini B, Camardi P, Silvestro L, Lace R, Bianchi M, The incidence of associated urological abnormalities in children with renal ectopia. J Urol 2004;172:1757‑1759. [ Links ]
35. Yuksel A, Batukan C, Sonographic findings of fetuses with an empty renal fossa and normal amniotic fluid volume. Fetal Diagn Ther 2004;19:525‑532. [ Links ]
36. Odiase VO, Horseshoe kidney. A review of 25 cases. J R Coll Surg Edinb 1983;28:41‑45. [ Links ]
37. Koff SA, Mutabagani KH, Anomalies of the kidney. In: Adult and Pediatric Urology, 3rd ed, Gillenwater JY, Grayhack JT, Howards SS, Mitchell ME (Eds), Lippincott Williams and Wilkins, Philadelphia 2002. p.2129. [ Links ]
38. RIVUR Trial Investigators, Hoberman A, Greenfield SP, Mattoo TK, Keren R, Mathews R, Pohl HG, Kropp BP, Skoog SJ, Nelson CP, Moxey‑Mims M, Chesney RW, Carpenter MA, Antimicrobial prophylaxis for children with vesicoureteral reflux. N Engl J Med 2014;370:2367‑2376. [ Links ]
39. Lee RS, Cendron M, Kinnamon DD, et al. Antenatal hydronephrosis as a predictor of postnatal outcome: a meta‑analysis. Pediatrics 2006;118:586‑593. [ Links ]
40. Rosen S, Peters CA, Chevalier RL, et al. The kidney in congenital ureteropelvic junction obstruction: a spectrum from normal to nephrectomy. J Urol 2008:179:1257‑1263. [ Links ]
41. Chertin B, Pollack A, Koulikov D, et al. Conservative treatment of uretero‑pelvic junction obstruction in children with antenatal diagnosis of hydronephrosis: lessons learned after 16 years of follow‑up. 2006. Eur Urol 49:734‑738.
42. Mei H, Pu J, Yang C, et al. Laparoscopic versus open pyeloplasty for ureteropelvic junction obstruction in children: a systematic review and meta‑analysis. J Endourol 2011: 25:727‑736 [ Links ]
43. Cussen LJ, Dimensions of the normal ureter in infancy and childhood. Invest Urol 1967;5:164‑178. [ Links ]
44. Stoll C, Alembik Y, Roth MP, Dott B, Sauvage P, Risk factors in internal urinary system malformations. Pediatr Nephrol 1990;4:319‑323. [ Links ]
45. McLellan DL, Retik AB, Bauer SB, et al. Rate and predictors of spontaneous resolution of prenatally diagnosed nonrefluxing megaureter. J Urol 2002;168:2177‑2180. [ Links ]
46. Brown T, Mandell J, Lebowitz RL, Neonatal hydronephrosis in the era of sonography. AJR Am J Roentgenol 1987;148:959‑963. [ Links ]
47. Krishnan A, de Souza A, Konijeti R, et al. The anatomy and embryology of posterior urethral valves. J Urol 2006;175:1214‑1220. [ Links ]
48. DeFoor W, Clark C, Jackson E, Reddy P, Minevich E, Sheldon C, Risk factors for end stage renal disease in children with posterior urethral valves. J Urol 2008;180:1705‑1708. [ Links ]
49. Holmes N, Harrison MR, Baskin LS, Fetal surgery for posterior urethral valves: long‑term postnatal outcomes. Pediatrics 2001;108:1‑7. [ Links ]
50. Sarhan O, Zaccaria I, Macher MA, et al. Long‑term outcome of prenatally detected posterior urethral valves: single‑center study of 65 cases managed by primary valve ablation. J Urol 2008;179:307‑312. [ Links ]
51. Ghanem MA, Wolffenbuttel KP, De Vylder A, et al. Long‑term bladder dysfunction and renal function in boys with posterior urethral valves based on urodynamic findings. J Urol 2004;171:2409‑2412. [ Links ]
52. DeFoor W, Clark C, Jackson E, et al. Risk factors for end‑stage renal disease in children with posterior urethral valves. J Urol. 2008;180:1705‑1708. [ Links ]
53. Van Eerde AM, Meutgeert MH, de Jong TP, Giltay JC, Vesico‑ureteral reflux in children with prenatally detected hydronephrosis: a systematic review. Ultrasound Obstet Gynecol 2007;29:463‑469. [ Links ]
54. Skoog SJ, Peters CA, Arant BS Jr, et al. Pediatric Vesicoureteral Reflux Guidelines Panel Summary Report: Clinical Practice Guidelines for Screening Siblings of Children With Vesicoureteral Reflux and Neonates/Infants With Prenatal Hydronephrosis. J Urol 2010;184:1145‑1151. [ Links ]
55. Hoberman A, Charron M, Hickey RW, Baskin M, Kearney DH, Wald ER, Imaging studies after a first febrile urinary tract infection in young children. N Engl J Med. 2003;348:195‑202. [ Links ]
56. Willemsen J, Nijman RJ, Vesicoureteral reflux and videourodynamic studies: results of a prospective study. Urology 2000;55:939‑943. [ Links ]
57. Cordell HJ, Darlay R, Charoen P, et al, UK VUR Study Group, Whole‑genome linkage and association scan in primary, nonsyndromic vesicoureteric reflux. J Am Soc Nephrol 2010;21:113‑123. [ Links ]
58. Metcalfe CB, Macneily AE, Afshar K, Reliability assessment of international grading system for vesicoureteral reflux. J Urol 2012;188:1490‑1492. [ Links ]
59. Upadhyay J, McLorie GA, Bolduc S, Bägli DJ, Khoury AE, Farhat W, Natural history of neonatal reflux associated with prenatal hydronephrosis: long‑term results of a prospective study. J Urol 2003;169:1837‑1841. [ Links ]
60. Sjöström S, Sillén U, Jodal U, Sameby L, Sixt R, Stokland E, Predictive factors for resolution of congenital high grade vesicoureteral reflux in infants: results of univariate and multivariate analyses. J Urol 2010;183:1177‑1184. [ Links ]
Disclosure of potential conflicts of interest: None declared
Received for publication: Sep 7, 2018
Accepted in revised form: Dec 7, 2018

