











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
BACKGROUND
In 2006 we implemented at Centro Hospitalar Universitário de Lisboa Central (CHULC) a Kidney Genetics Clinic (KGC) with an initial focus on autosomal dominant polycystic kidney disease (ADPKD). With the coming of age for novel ADPKD therapies, our main objectives were the following: i) gain expertise in the management of ADPKD patients (including molecular diagnosis); ii) gaining knowledge on total kidney volume as a biomarker; iii) develop a cohort that could be useful in future clinical trial settings.
In those days, the Sanger sequencing days, molecular diagnosis was not easily available in Portugal. In particular, the PKD1 gene was (and still remains) a very difficult gene to sequence. The idea was to develop molecular genetics skills in our Laboratório de Morfologia Renal and provide indirect genetic testing for ADPKD. The methodology relied on the co‑segregation analysis of the disease with several polymorphic microsatellite repeats in closed proximity (linked) to the causal gene. As expected, the limitations soon became apparent: the test required multigenerational families and needed as many as possible (affected and non‑affected) members available for segregation studies; and although in some families we could identify and trace the ancestral chromosome it did not allow us to identify the actual mutation, the purpose of the whole thing being hard to explain to colleagues and, most importantly, to families.
On the other hand, we successfully set up a computerized tomography (CT) scan‑based segmentation methodology for total kidney volume (TKV) determination. A pilot study was performed and the results presented in 20081. Finally, regarding clinical trials, and based on the literature at the time, an Investigator Originated Proposal (IOP) entitled “National Multicenter Sirolimus (SRL) Evaluation Trial for Autosomal Dominant Polycystic Kidney Disease (ADPKD): a new use for an old drug?” was submitted to the Wyeth Pharmaceuticals. Soon after the IOP submission, Pfizer acquired Wyeth and later on it became apparent that SRL was not effective in patients with ADPKD2.
The absence, at both CHULC and the medical school we are affiliated to, of a molecular genetics diagnostic laboratory has been major limitation to our program. This was one of the reasons we restricted our approach to linkage analysis in ADPKD, the most prevalente inherited kidney disease, since it was the only molecular approach we could afford. Apart from ADPKD, the other mendelian traits we performed molecular diagnosis concerned the identification of the NPHP1 “common” deletion in homozigocity for Nephronophthisis (NPH), using reagents that the corresponding author had brought along from his PhD studies3, the direct sequencing of the coding region of the SLC5A2 gene in Familial Renal Glucosuria (FRG), a major investigational subject for the authors4, as well for the UMOD gene in cases of Familial Juvenile Hyperuricemic Nephropathy (FJHN), for which research funds were also available.
With the growing awareness of the clinic’s activity, an increasing number of adult patients with other inherited kidney diseases was being referred. Most importantly, the widespread use of next generation sequencing (NGS) made commercial gene‑panel testing widely available and affordable. In light of these developments the scope of our activity broadened and now encompasses a wide range of inherited renal diseases. For these, expertise management is provided, including phenotype‑driven panel testing outsourced to commercial as well as academic institutions. It is the purpose of the current manuscript to review the KGC’s activity for the past 5 years. We will emphasize the impact of genetic testing in patients but also address the role of renal imaging and target pharmacological interventions already available for some inherited renal diseases.
MATERIAL AND METHODS
We reviewed the clinic’s activity from the 1st of January of 2016 until the 31st of December of 2020, and selected the following outcomes:
i) number, gender and age of individuals referred and number of appointments;
ii) clinical diagnosis, genetic tests requested and the reported results by gene;
iii) number of specific imaging procedures requested: TKV for ADPKD and angiomyolipoma (AML) volume assessment for the Tuberous Sclerosis Complex (TSC);
iv) number of individuals selected for specific therapeutics.
Phenotypes were named after the Orphanet, assessed via the portal site (https://www.orpha.net), which allows codes for phenotypes, as opposed to on‑line Mendelian Inheritance in Man (OMIM) (https://omim.org) that is often gene‑ontology based for the more genetically heterogenous clinical traits (e.g. ADPKD). Disease definition followed formal diagnostic criteria whenever available: ADPKD5, Alport Syndrome (AS)6, Autosomal Dominant Tubulointerstitial Kidney Disease (ADTKD)7, atypical Hemolytic Uremic Syndrome (aHUS)8, TSC9, FRG4, Gitelman Syndrome (GS)10 and Bartter Syndrome (BS)11. Also, and regarding the adult population with Genetic Steroid Resistant Nephrotic Syndrome (gSRNS, ORPHA code 656), focal and segmental glomerulosclerosis (FSGS) is the most frequent histopathological finding. Likewise, and in contrast to the pediatric setting, autosomal dominant forms prevail. As such, we have included probands from pedigrees having familial cases (even if steroid resistance was not assessed) as well those seemingly sporadic FSGS young adult cases displaying primary resistance to steroids. In all circumstances the histopathology of FSGS had to be documented: if not in the proband, at least in an affected family member. For that reason, we opted for the denomination of FSGS instead of gSRNS.
It is our intention only to report pathogenic or likely pathogenic allelic variants, according to the American College of Medical Genetics and Genomics12. But we must acknowledge that these guidelines have not always been around and so it is possible that variants of unknown significance may have been included for earlier cases. TKV, as a biomarker for ADPKD, was assessed by a low dose radiation (5‑10 mSv) CT scan protocol with measurement of each kidney’s sagittal and coronal lengths as well width and depth by a single radiologist experienced with ADPKD. Height adjusted TKV (ht TKV) was determined using the ellipsoid formula (π/6xLxWxD) and the Mayo clinic imaging classification algorithm for ADPKD (https://www.mayo.edu/research/documents/pkd‑center‑adpkd‑classification/doc‑20094754).
For AML evaluation in TSC and patient selection for the treatment with Everolimus/Votubia™ or follow‑up, a protocol using Magnetic Resonance Imaging (MRI) was develop where up to 5 lesions are selected, the volume of each determined by ellipsoid method (3 orthogonal measures for each AML) at week t0, t12, t48 and annually thereafter. As we gained experience with MRI for monitoring AML, the protocol was changed and MRIs are now performed at week t0, t24 and annually after.
Finally, and concerning targeted and disease specific therapies for inherited renal disease already provided by CHULC/KGC, we have reviewed the data for patients that were selected and have initiated therapy with Everolimus/Votubia™ (TSC), Eculizumab/Soliris™ (aHUS) and Tolvaptan/Jinarc™ (ADPKD).
RESULTS
Structure of the kidney genetic clinic
A single nephrologist with an interest in inherited renal diseases and with formal training in molecular genetics is responsible for the appointments as well all the necessary bench work. In addition, residentes from the genetic and nephrology fellowships have joined the program for periods of 3 months as part of their postgraduate training.
A part‑time administrative support is also available.
Two weekly dedicated genetic clinic sessions are scheduled, encompassing 8‑10 appointments per week and accounting for a total of ~10 hours. An additional 2 hours are allocated to sample collection, DNA extraction, storage and submission to outsourced genetic testing sites, contacting laboratories for results discussion and panel optimization, as well as other administrative work.
A recent and important feature of the KGC is the multidisciplinar rounds between adult and pediatric nephrologists and a geneticista that take place every other week to discuss the most difficult cases, update the genetic testing results and develop multigene panel for NGS sequencing.
Figure 1 depicts the structure of KGC. Although occasional patients are referred from other hospitals, the majority are from within CHULC, in particular from the pediatric and adult nephrology departments.
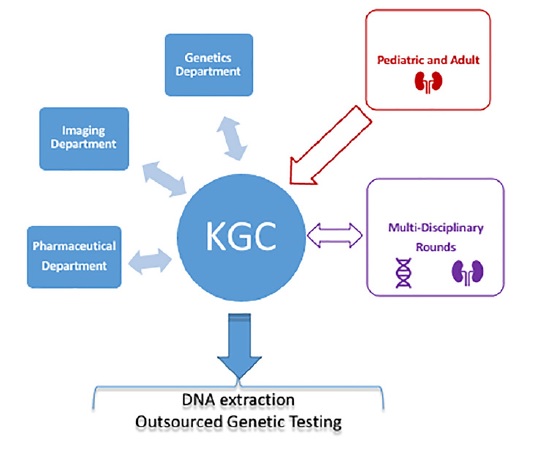
The KGC relies on the genetic department of CHULC for formal genetic counselling and so all the genetic tests performed for KGC patients in that context are ordered by the genetics department. Also, many pediatric patients that have transitioned to the KGC have already been genotyped. The large majority of the genetic testing requested by KGC concerns diagnostic molecular confirmation in cases with atypical presentations, sporadic forms or if a phenocopy is suspected, as well when genotyping can help prognostication and patient selection for targeted therapies (Tolvaptan/Jinarc™ and living related donor organ transplantation).
Whenever possible, the most suitable proband within the affected family is the individual to be selected for genetic testing. This will be relevant in cases of X‑linked Alport syndrome, where hemizygotic male patients with fully penetrant disease should be tested first, or in the adult setting of FSGS, where patients with a renal biopsy are
eligible. Once the patient has given the informed consent, a blood sample is collected and DNA extracted from peripheral blood leucocytes that will be stored at ‑20ºC until a statement of responsibility is issued by the board of CHULC. Only then will the sample be sent to the commissioned genetic laboratory by mail. Extracting DNA in our own lab is most advantageous for patients since collecting a sample during the routine appointment will avoid a second visit once the statement of responsibility is issued. In addition, if there is need for a second round of testing for a different set of genes (either because novel genes were meanwhile found to be responsible for the phenotype or the latter was rechallenged by novel clinical data) the stored whole peripheral blood enables additional rounds of DNA extraction.
Referrals, appointments and epidemiological data
During the study period, 293 individuals were referred to the KGC. The average age and the number of appointments per patient were 46.8 years (± 16.5) and 5.3 (± 4.5), respectively, with 55.4% of the individuals being females.
Clinical and Genetic diagnosis
Table 1 details the clinical diagnosis, gender and age for the entire KGC cohort as well for each of the 10 most prevalent observed inherited kidney disorders. For each diagnosis, the corresponding Orphanet code is displayed. In the category of “Others” there are 19 referrals that include genetically characterized cystic phenotypes [eg, Bardet‑Biedl Syndrome (1) or Nephronophthisis (NPH) (1)] as well nephrolithiasis cases (3) that remained uncharacterized after phenotypic evaluation and for whom the caring physician pursed a genetic cause.
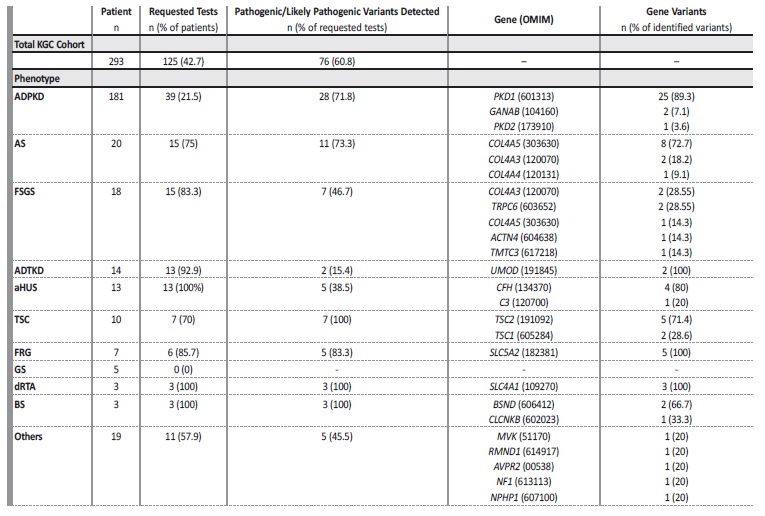
In Table 2, and for each clinical diagnosis, the number of genetic tests performed as well as their results assorted by gene are presented. For the entire KGC cohort of the 293 individuals censored, 42.7% had a genetic test performed, adding to a total of 125 individuals tested. For 76 of these (60.8% of tested individuals), pathogenic or likely
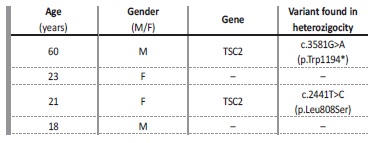
pathogenic alleles were identified and found to account for the phenotype. In particular, for recessive traits like Lissencephaly‑8 syndrome (1), Hyper‑IgD syndrome (1), Combined Oxidative Phosphorylation Deficiency‑11 (1), BS (3), NPH (1) and AS of autosomal recessive inheritance (1) there was a requirement for the discovery of 2 pathogenic alleles in TMTC3, MVK, RMND1, BSND, CLCNKB, NPHP1 or COL4A3 genes, respectively. In the current analysis, each pair of alleles found in these recessive phenotypes was computed as single entry.
Table 1 Demographic data of the KGC cohort.
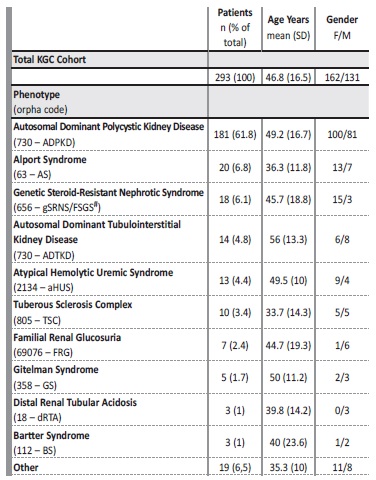
*classification by clinical diagnosis according to Orphanet; # see text for details
Imaging Procedures
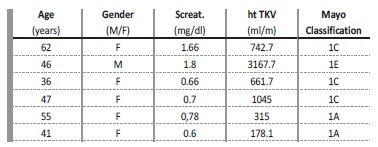
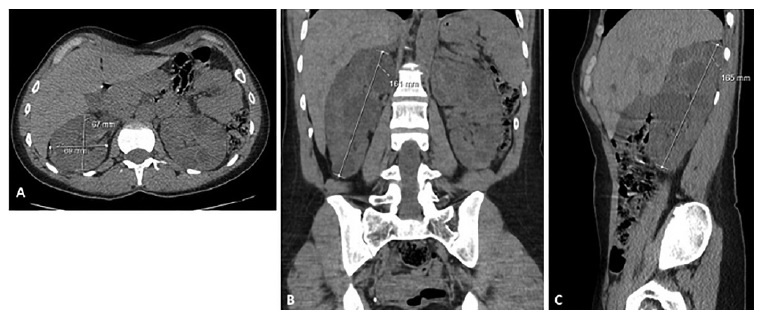
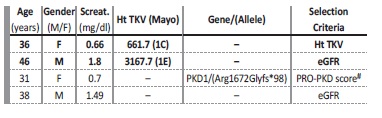
Ht TKV as a biomarker in ADPKD for renal prognostication and/or patient selection for Tolvaptan/Jinarc™ therapy was requested in 4 patients. In two additional patients, CT scans performed in diferente setting (liver transplantation, hemorrhagic or infected cysts) also enabled ht TKV determination. Patients’ age, gender, and serum creatinine for these 6 patients are presented in Table 3. As an example, Figure 2 displays the length measurements of the right kidney for the female patient aged 36, where axial, coronal and sagittal views are portraited.
For TSC, 3 individuals were assessed with MRI regarding AML. Figure 3 depicts an illustrative patient in whom our MRI protocol monitored 3 AML lesions before, during and after Everolimus/Votubia™ treatment.
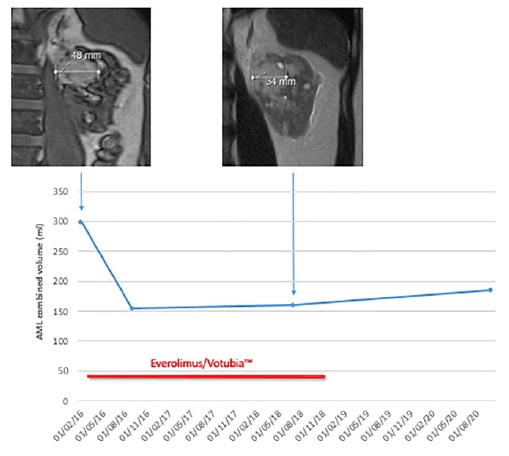
Figure 3 AML combined volume evolution in a patient before, during and after EVR treatment. Illustrative MRI findings are included
Disease specific and targeted therapies
Everolimus/Votubia™ - in 2016 we treated our first TSC patient for AML. Since then, and until the 31st of December 2020, 3 additional patients were recruited, including transitioning pediatric patients. Table 4 summarizes our treated TSC cohort. Eculizumab/Soliris™ - 3 patients with aHUS were treated with anti C5 therapy during the study period. All had histopathologically documented thrombotic microangiopathy by means of a renal biopsy, all were NGS sequenced using a multigene panel targeting the alternative complement pathway system and all have had previous relapses. Table 5 depicts the major findings of the 3 individuals.
Tolvaptan/Jinarc™ - V2 receptor antagonist therapy is the most recent disease specific therapy provided to CHULC patients. In 2020 we developed a protocol for Tolvaptan/Jinarc™ treatment of ADPKD patients that incorporated the available guidelines (see discussion) and we screened our cohort for ADPKD individuals for
treatment eligibility. The CHULC board approved treatment for the first 4 patients to be treated in 2021 and, at the time of this writing, 2 have already initiated treatment with Tolvaptan/Jinarc™. In Table 6 we report the phenotype and genotype (when available) in all 4 selected patients.

DISCUSSION
In the current analysis, we evaluated the KGC activity for the last 5 years. The range of phenotypes evaluated are in line with what has been reported by Lundquist AL et al.13 while describing a similar clinic at the Massachusetts General Hospital (Boston, USA) but with a cohort that is significantly smaller than ours. With the advent of NGS it became possible for institutions not associated with genetic testing laboratories to provide molecular genetic diagnosis for their patients in a manner independent of research funding availability.
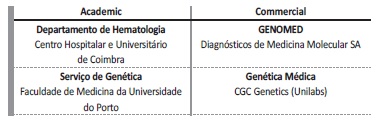
In Table 7 we provide a list of the most frequently outsourced academic and commercial laboratories that provide genetic testing to our patients.
As expected for an adult population, the cohort is mostly comprised (60.8%) of ADPKD individuals. Genetic testing for ADPKD is seldom requested in clinical practice and, accordingly, only 21.5% of our ADPKD patients underwent molecular diagnosis. Also, our detection rate of pathogenic/likely pathogenic variants is 71.8%, not different from that reported in the literature14. Collagen type IV‑related nephropathies account for the second largest group of individuals evaluated. They presented either as AS (n=20) or as an FSGS phenocopy2, with a high rate of pathogenic/likely pathogenic variant detection rate (73.3% for AS). Interestingly, for AS, the female gender predominates. Although some autosomal forms may account for this, we have observed that even for X‑linked AS pedigrees, more female (n=6) than man (n=2) were being tested.
One possible explanation is that sisters, daughters or mothers of affected male patients were being evaluated due, for instance, to hematuria. The third most frequent phenotype in our list is ADTKD, with a mutation detection of 15.4%. KGC ADTKD patients are part of our single‑center ADTKD registry that includes 25 individuals and has a similarly low rate of variant detection. For the interested reader, these are all included in the international UMOD Mutation Registry (http://j.mp/2q7Fi8f)15. As previously mentioned, in earlier years, we only sequenced the UMOD gene in FJHN pedigrees. More recently, a broader definition of ADTKD was introduced to accommodate the finding of loci heterogeneity and we are now able to request for REN, SEC61A and HNF1B (including HNF1B major DNA rearrangements). These 3 genes, however, account for a small minority of cases. Other than UMOD, reported in the literature to be responsible for 38.4% of ADTKD families, the other single relevant gene is MUC1, with a cytosine insertion occurring in the VNTR (variable number tandem repeats) of its coding region found in 35.1% of the ADTKD‑UMOD negative pedigrees16. Detection of the MUC1 insertion remains technically challenging and should be only pursued after having first sequenced UMOD. The 13 KGC patients with aHUS are also part of a larger single center registry that includes 41 patients and the observed 38.5% mutation detection rate is not different from the found for the whole registry (32%), but much lower than the 50‑60% figure generally reported8. The multigene panel tested in use already includes 11 genes coding for proteins regulating the alternative complement pathway. Contrary to ADTKD, in which a family history is a major diagnostic criterion, in aHUS the family history is rarely (if ever) present. Also, the registry of CHULC of adult aHUS patients is gene test driven and includes patients who were evaluated in the pre‑transplant setting with chronic kidney disease (CKD) of unknown etiology but in whom, and retrospectively, thrombotic microangiopathy (TMA) could not have been excluded and were, therefore, eligible for genetic screening. Realistically, only a fraction of those would have had TMA and even though, probably secondary in its nature. The identification of pathogenic/likely pathogenic variants in relevant genes enables the nephrologist to “molecularly” establish a diagnosis. From our experience, this turned out to be relevant in the following circumstances:
i) Typical phenotypes with atypical patterns of inheritance (e.g., autosomal AS);
ii) Atypical/incomplete phenotype expression in sporadic cases of a mendelian trait (e.g., de novo ADPKD);
iii) A substitute for renal biopsy in tubulo‑interstitial disease with or without family history (e.g., ADTKD, NPH);
iv) Ascertaining the correct etiology in face of non‑specific histopathological findings (e.g., FSGS);
Overlapping with these and in the particular setting of renal transplantation, an informative genetic test was relevant because it:
v) Enabled living related kidney donation (e.g., NPH);
vi) Correctly assessed the risk of disease recurrence (e.g., genetic forms of FSGS (no risk), aHUS (high risk).
The Pharmaceutical Department of CHULC can now provide Tolvaptan/Jinarc™ for our ADPKD patients. Tolvaptan/Jinarc™ was approved for the treatment of rapidly progressive forms of ADPKD following the TEMPO trials17,18. European (eGFR centered) and American (ht TKV based) guidelines are available19,20and they provided the frame for the treatment protocol we have implemented in CHULC (available on request). Tolvaptan/Jinarc™ is the first approved medicine for ADPKD. Novel therapies are also emerging and the future looks bright for upcoming ADPKD generations.
Unfortunately, the future also comes with an expensive price tag. ADPKD is not a rare disease. Using a population sequencing methodology, prevalence estimates are of 9.3/1000021. If one considers the population for which CHULC provides nephrology care (360000 in Lisbon + 250000 in Vila Franca de Xira) one can assume that there are 567 individuals with ADPKD in that population alone. Selecting and providing specific therapies for the ADPKD population will cause a heavy burden on nephrology departments because referrals must now occur long before serum creatinine rises. In addition to the medicine cost, one has to anticipate the imaging procedures as well a more widespread use of genetic testing that will be needed for proper risk stratification. Finally, if the patient is selected for treatment, monthly visits are scheduled for the first 1.5 years of treatment, in striking contrast to the current practice of every‑other‑year appointment or no appointments at all (for those with normal serum creatinine). We have already started to prioritize patients for Tolvaptan/Jinarc™ treatment approval in 2022. The initial feverish enthusiasm of getting our first patients treated in 2021 has given place to ethical concerns on who will not get it in next coming years. Importantly, and aware that our views may not be representative of the nationwide nephrology community, it is imperative that we reach a national consensus for ADPKD current and future treatments, in order to prevent discrepancies in patients’ access to therapy.
Our study has several limitations. It did not evaluate the costs of genetic testing or those related to disease specific medicines. Also, we have not detailed the turnaround times for genetic testing.
Finally, by characterizing patients and not pedigrees, we probably overestimated the prevalence of some rare phenotypes and alleles, and incurred in gender distribution bias, as already mentioned for X‑linked AS. But regardless of the above‑mentioned limitations, this analysis enabled us to assume the following considerations. First, that even in the absence of an affiliated genetic laboratory, it is still possible to provide patients with molecular genetics testing. Second, that patients are best served by physicians who see a high volume of cases like theirs, this being particularly true when it comes to rare and orphan diseases. In fact, deploying precision medicine tools (diagnostic as well therapeutics) was only possible because of the KGC enrichment nature of the referral. Third, that genetic testing performed outside formal genetic counselling or research scopes ought to primarily concern cases that remain unsolved after non‑invasive clinical/phenotype evaluation. This will allow us, in the future, to focus on novel approaches like whole exome sequencing as exemplified by a recent report that established a mendelian cause in almost 10% of CKD cases of unknown etiology22. Lastly, that collaborative work between pediatric and adult nephrologists sharing a common interest in inherited kidney disorders is the best way to approach familial disorders and ensure proper continuity of treatment for pediatric patients.
In the adult nephrology environment, molecular genetics only seldom impacts on formal genetic counselling. Still, and as hereby exemplified, genotype information often provides the best way nephrologists can reach precision diagnostics and personalized therapeutics.
There are 2 additional areas for which genetics and Genomics are revolutionizing nephrology. The first one concerns the more prevalente complex/multifactorial traits such as the African‑American populations’ susceptibility to CKD, where APOL1 alleles play a major role23.
The other being gene therapies, like those based on siRNA platforms, in particular Lumasiran, an siRNA already approved for patients with Primary Hyperoxaluria, but only for those displaying pathogenic variants in the AGXT gene24.
Transitioning from the 40‑year‑old Evidence Based Medicine into modern day Precision Medicine (patient‑centered) will only be possible if we become knowledgeable about Genomics concepts and lexicon. For the next generation of nephrologists, understanding the Genomic basis of kidney diseases (either monogenic or complex/ multifactorial in their nature) will be more relevant than the study of renal histopathology. Revising and updating nephrology fellowship curricula is mandatory in order to overcome present‑day medical illiteracy on this subject. If we want our patients to have a glimpse of the future, we must stop looking to the past. The time has come to move forward.

