











 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
INTRODUCTION
Hemolytic uremic syndrome (HUS) is a thrombotic microangiopathy (TMA) characterized by hemolytic anemia, thrombocytopenia and acute kidney injury. HUS can be caused by several conditions: shiga toxin, Streptococcus pneumonia infection, complemente dysregulation, mutation of diacylglycerol kinase ɛ, cobalamin C defect or secondary HUS (infections, drugs, cancer and systemic diseases).1
Atypical HUS (aHUS) is due to presumed or confirmed genetic or acquired dysregulation of the complement alternative pathway, which is detected in 40%-60% of patients.1 Sixty to 70% of aHUS patients carry identifiable mutations in complement genes or anticomplement factor H (CFH) antibodies that result in loss of protection of endothelial cells and platelets from complement attack.2 Mutations in 6 genes have been associated with increased susceptibility for aHUS (CFH, CFI, MCP, C3, CFB and thrombomodulin - THBD) and should be analyzed by direct sequencing. Combined mutations are found in 3%-6%.2
Many triggers are known to induce aHUS, including infections by Epstein-Barr virus, varicella and human immunodeficiency virus.3-7 It is still unknown whether patients with HUS triggered by infections (as well as drugs or other causes) also have mutations which are not yet described. HUS triggered by influenza virus (iHUS) is rare.8 In almost all cases it is associated with influenza A virus, mainly A(H3N2) and A(H1N1).8 Only few cases of HUS associated with influenza B vírus infection have been recently published, so these may be underestimated.2-4Genetic testing revealed later on in all patients an underlying genetic complement dysregulation.3,4 aHUS may be a serious life-threatening condition, progressing to end-stage renal disease in 50% to 80% of cases, depending on the underlying defect.3,5 Complement blockade using a monoclonal anti-C5 antibody, eculizumab, has greatly improved the outcome in recente years for certain groups of HUS, but not all.1,6
Due to its rarity, we describe an aHUS case with influenza B virus infection as a trigger, as well as the decision on whether treating or not with eculizumab. Genetic screening and vaccination will also be discussed.
CASE REPORT
During influenza season a 13-year-old boy presented with two days of flu-like syndrome (maximum temperature 38.8°C, cough, abdominal cramps and vomiting), then with dark-coloured urine and finally anuria.
He had irrelevant family history and two pneumonia episodes at the age of three. Although vaccinated in previous years, he missed fluvaccination in the present year. The patient presented with jaundice, blood pressure in the 95th - 99th + 5 mmHg centile (129/84 mmHg), petechial lesions on the right shoulder, abdomen and trunk and no meningeal signs. Analysis at the admission with re-evaluation few hours later (see Fig. 1 for analytical evolution) revealed hemolytic anemia, thrombocytopenia, renal failure and elevated bilirubin and LDH.
Coombs test was negative and a blood smear showed rare schistocytes. The urinalysis showed proteinuria (3+) and granular casts and renal ultrasound revealed enlarged hyperechogenic kidneys. ADAMTS13 activity was 0.79 (RR ≥0.67). The results established the diagnosis of HUS. Complement activation could be confirmed by mildly reduced serum C3 (0.81 g/L; RR 0.90-1.08 g/L), with normal serum C4 (0.27 g/L; RR 0.16-0.48 g/L). Complement factor B (CFB) was slightly elevated (54.5 mg/dL; RR 17.04-51.06), factor H (CFH) was normal (54.8 mg/dL; RR 47.45-65.99), factor I (CFI) was low (2.27 mg/dL; RR 2.38-3.18) and anti-CFH antibodies were practically normal (32.61 UA/mL; RR<27). The complement alternate pathway (AH50) assay was 112% (RR>70%). Assays were negative for antinuclear antibody (ANA), anti-β2-glycoprotein I antibodies IgG/IgM, antineutrophil cytoplasmic antibodies (ANCA), lupus anticoagulant and anticardiolipin antibodies.
No pathogenic enterobacteria (enterohemorrhagic E. coli, Shigella, Yersinia or Campylobacter species) were detected by culture as well as no Shiga toxin by PCR in multiple stool samples. A nasopharyngeal swab was positive for Influenza B DNA by multiplex-PCR and negative for Group A Streptococcus antigen. HBs antigen, hepatitis C antibody and anti-HIV 1 and 2 antibodies were all negative. Urine samples were negative for pneumococcal urinary antigen test.
The patient was admitted in day one to Intensive Care Pediatric Unit due to anuria and analytical deterioration. He showed favorable clinical and analytical evolution (Fig. 1), being transferred to Nephrology Unit at day four. Progressive normalization of renal function was noticed, with normal blood pressure and diuresis. Treatment was rendered for five days with oseltamivir and cefotaxime.
At this point, secondary HUS to Influenza B infection or aHUS due to complement mutation with Influenza B virus as a trigger were considered as diagnostic hypothesis. Subsequently, genomic analyses of patient’s complement including 11 genes (CFH, CD46 (MCP), CFI, C3, THBD, CFB, CFHR5, CFHR1, CFHR3, CFHR5, DGKE) revealed a pathogenic heterozygotic missense variant on CD46 (MCP) gene, c.554A>G, p.Asp185Gly. CHFR1 and CHFR3 mutations were not detected.
As Influenza B had been identified and oseltamivir was prescribed and both clinical and analytical course were favourable, treatment with eculizumab was not performed and patient was kept under close clinical and laboratorial follow-up. He was also immunized with quadrivalent conjugate meningococcal (MenACWY-TT), meningococcal B (4CMenB) and hepatitis B vaccines, in the event of a recurrence, in which case treatment with eculizumab should be indicated. He was kept under close clinical and laboratorial follow-up at outpatient setting for several months, with hematological and renal function normalization. Anti-factor H antibodies were repeated one month after first titration and showed a similar almost normal value. Presently, he is under annual follow-up without any signs of relapse; annual influenza vaccine is administrated seasonally, without adverse effects.
DISCUSSION
iHUS is rare and mostly associated with Influenza A infection. There are only few reports of aHUS triggered by Influenza B, all identified cases associated with mutations in the MCP or C3 gene, occasionally combined with other mutations.3,4,8The outcome is generally favorable, depending on underlying complement gene deficiencies and/or the presence of CFH or ADAMTS13 autoantibodies.8 Differentiation between HUS due to complement dysregulation with influenza virus as a trigger and influenza-associated HUS without identifiable complement abnormalities, is critical for acute and long-term management.8
Patients with MCP-mutations, as on this case, often show spontaneous remission and present a lower risk to progress to end-stage renal disease than patients with other mutations in genes for regulators for the alternative pathway complement system (e.g., factor H, factor I).4,9
Further delineation of risk haplotypes, specific microbial agents or their products could be interesting concerning therapeutic and prevention.8
All patients should be genetically characterized for confirmation on complement-dependency or not; establishing prognosis, risk of relapses and progression to ESRD; genetic counselling to parents and family; decisions for kidney transplantation; and eventual complement blockade treatment discontinuation (further prospective studies required).2
Incomplete penetrance of aHUS in carriers of mutations is common to all aHUS-associated complement genes and it is now established that the overall genetic predisposition to aHUS of an individual results from the combination of different inherited factors.10 Not all patients with genetic susceptibility will develop the disease, with an environmental factor that triggers the complement cascade being needed for disease to occur.10 Influenza B might have pathogenic effects similar to influenza
A in triggering aHUS,4 whether through an altered immune response with activation of mononuclear cell which produces tumor necrosis factor alpha, or exposing the Thomsen-Friedenreich antigen on red blood cells, platelets and glomerular epithelia caused by viral neuraminidase that cleaves sialic acid residues of several glycoproteins.3,11 12
MCP (CD46) serves as a cofactor for factor I to mediate inactivation of C3b and C4b deposited on host cells. Several disease-associated mutations (rheumatoid arthritis, asthma, etc.) in MCP and some putative links associated to different pathologies (systemic lupus erythematosus, glomerulonephritis, and pregnancy-related disorders) have been identified.13 Mutations in MCP may also cause some susceptibility to infections. CD46 activation by pathogenic microorganisms induces a rise of interleukin-2 (IL-2) secretion and, after several intermediate steps, the generation of an IFN-γ−/IL-10+T cell self-regulatory phenotype (Treg). Thus, complement-induced Tregs provide a supportive role by facilitating B-cell activation and as the immune response progresses, complement-induced Tregs might then control the immune response.13 Consequently, disruption of these pathways may contribute to vulnerability to infections.13 In our case, episodes of pneumonia were also reported, prior to the Influenza B infection.
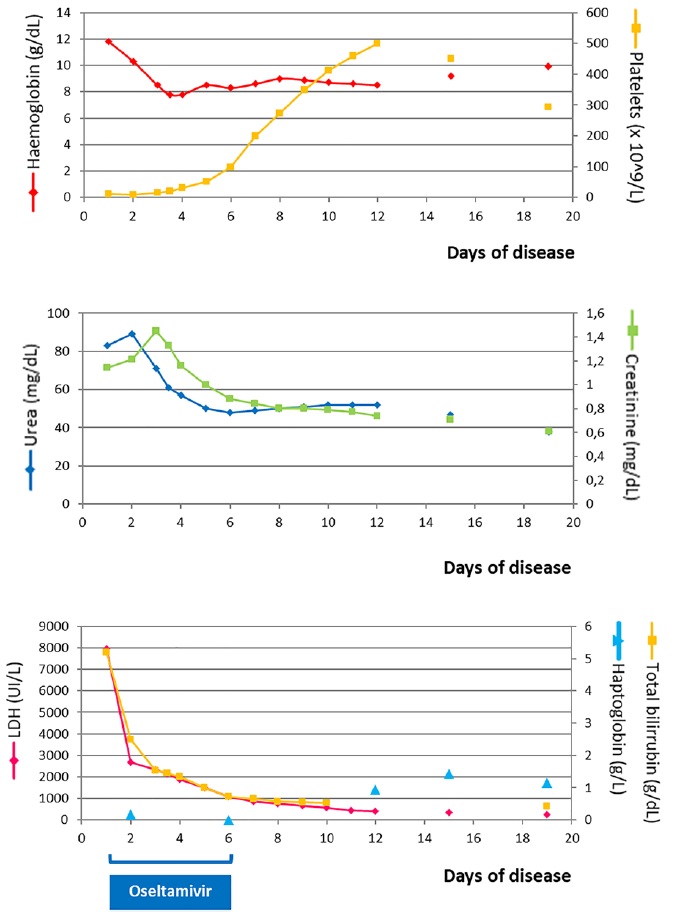
For aHUS in children eculizumab is proposed as first-line treatment.2 While the treatment is effective in patients with or without a confirmed complement mutation, genetic screening is required for the longer-term management. Anti-CFH antibody testing is the only complement investigation urgently required during the acute phase, as a positive result implies alternative treatment options.7 Since our patient presented a favourable clinical and analytical course and a heterozygotic missense variant on CD46 (MCP) gene was detected in molecular study, treatment with eculizumab was not performed and the patient was kept under close clinical and laboratorial followup.
However, it should be considered in case of relapse.2,4 Anti-factor H antibodies, despite positive, were of very low level; also, CHFR1 and CHFR3 mutations, strongly associated with anti-factor B antibodies presence, were not detected. Taking this into account, and the fact that their second titration showed a similar level and specially the very favorable evolution of the patient, we decided not to prescribe immunosuppression.
Data concerning immunization history is scarce on iHUS case reports.8 Few reports associate vaccination to HUS or TTP8, but data are limited and should not constitute evidence against active immunization. The high variability of the predicted antigen changes also hampers the efficacy of influenza vaccines.8 The aHUS registry or other HUS databases might be able to retrospectively analyze information on safety and efficacy of influenza vaccination8,14 and prospective studies addressing influenza vaccination strategies in aHUS patients would be important to optimize care in this subpopulation.8 In our patient we decided to administrate annual influenza vaccine since he already took it in previous years without adverse effects.
In conclusion, aHUS patients should be screened for all known diseaseassociated genes. Screening should not be stopped after finding a mutation, to avoid missing other genetic susceptibility factors influencing gene phenotype, particularly in patients with MCP or CFI mutations.
The decision on whether treating or not with eculizumab should be made based on clinical and laboratorial evolution as well as molecular studies results. Influenza B is a trigger for aHUS and might be underreported as such. Influenza vaccination may protect patients at risk.

