










Servicios Personalizados
Revista

Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
 Citado por SciELO
Citado por SciELO Links relacionados
 Similares en
SciELO
Similares en
SciELO Compartir

 Permalink
PermalinkNascer e Crescer
versión impresa ISSN 0872-0754versión On-line ISSN 2183-9417
Nascer e Crescer vol.28 no.3 Porto set. 2019
https://doi.org/10.25753/BirthGrowthMJ.v28.i3.14159
CASE REPORTS | CASOS CLÍNICOS
Retroperitoneal paraganglioma, a rare cause of arterial hypertension
Paraganglioma retroperitoneal, uma causa rara de hipertensão arterial
Mariana BrancoI, Francisca MartinsI, Ariana TelesI, Vera GonçalvesI, Francisco Ribeiro-MourãoI, Idalina MacielI, Ana CarneiroI
I - Department of Pediatrics, Hospital de Santa Luzia, Unidade Local de Saúde do Alto Minho. 4901-858 Viana do Castelo, Portugal. mariana.a.branco@hotmail.com; franciscamartins@hotmail.com; arianajbt@gmail.com; vgveragoncalves@gmail.com; franciscoribeiromourao@gmail.com; idalina.maciel@gmail.com; anacatfcsousa@gmail.com
Endereço para correspondência | Dirección para correspondencia | Correspondence
ABSTRACT
Introduction: Paragangliomas are rare tumors in pediatric age, derived from sympathetic tissue of extra-adrenal location or parasympathetic tissue. Some of these tumors secrete catecholamines, being a rare cause of hypertension.
Clinical Case: A female adolescent, with 14 years old and unremarkable medical history, was referred to the Pediatric consultation due to episodes of holocranial headache associated with blurred vision with six months of evolution. Physical examination and brain magnetic resonance imaging revealed no abnormalities. Physical examination showed stage 1 hypertension, later confirmed by ambulatory blood pressure measurement. Analytical study, chest X-ray, electrocardiogram, and echocardiogram were normal. Renal echography revealed a heterogeneous mass contiguous to the left renal sinus and abdominal computed tomography showed a retroperitoneal lesion in the medial side of the left perirenal space. Due to retroperitoneal paraganglioma suspicion, the patient was referred to Porto’s Portuguese Institute of Oncology, where surgical excision of the lesion was performed. Anatomopathological examination confirmed diagnosis of sympathetic paraganglioma.
Conclusion: Although rare in pediatric age, paragangliomas represent the most common endocrine tumor. Biochemical and imaging evaluation are key for diagnosis, after which a genetic study is recommended due to its relevance for patient management and follow-up. Treatment consists in surgical excision, and complete remission is achieved in approximately 90% of cases.
Keywords: catecholamine; arterial hypertension; paraganglioma
RESUMO
Introdução: Os paragangliomas constituem tumores raros em idade pediátrica, que derivam do tecido simpático de localização extra supra-renal ou do tecido parassimpático. Alguns destes tumores segregam catecolaminas e são uma causa rara de hipertensão arterial.
Caso Clínico: Uma adolescente do sexo feminino, de 14 anos de idade e sem antecedentes pessoais de relevo, foi referenciada à consulta de Pediatria por episódios de cefaleia holocraniana associados a visão turva com seis meses de evolução. O exame físico e a ressonância magnética cerebral encontravam-se normais. Na consulta foi detetada hipertensão arterial estadio 1, confirmada através de monitorização ambulatória da pressão arterial. O estudo analítico, radiografia de tórax, eletrocardiograma e ecocardiograma não mostraram alterações. A ecografia renal revelou uma massa heterogénea em contiguidade com o seio renal esquerdo e a tomografia computorizada abdominal evidenciou uma lesão retroperitoneal na vertente medial do espaço perirrenal esquerdo. Perante suspeita de paraganglioma retroperitoneal, a doente foi encaminhada para o Instituto Português de Oncologia do Porto, onde realizou exérese cirúrgica da lesão, tendo o exame anatomopatológico confirmado o diagnóstico.
Discussão: Embora raros em idade pediátrica, os paragangliomas representam o tumor endócrino mais comum. Perante suspeita clínica de paraganglioma, o diagnóstico bioquímico e imagiológico é fundamental. Após o diagnóstico, é recomendada a realização de estudo genético para uma melhor abordagem e seguimento do doente. O tratamento consiste na exérese cirúrgica, sendo alcançada remissão completa em cerca de 90% dos casos.
Palavras chave: catecolaminas; hipertensão arterial; paraganglioma
Introduction
Paragangliomas are rare neuroendocrine tumors in pediatric age that arise from sympathetic tissue of adrenal or extra-adrenal location or parasympathetic tissue. Paragangliomas arising from adrenal chromaffin cells represent a specific subtype termed pheochromocytomas, accounting for 80−85% of cases.1-4
Although rare, with an incidence of 1:100.000 patients per year in the general population, paragangliomas represent the most common endocrine tumor in pediatric age.1,5 Approximately 10% of pheochromocytomas and 40% of paragangliomas are malignant.5 Although during childhood these tumors are more prevalent in males, this trend reverses in adolescence, probably due to hormonal influences.5
Paragangliomas may occur as sporadic or familial entities. Familial syndromes commonly associated with paragangliomas are Von Hippel-Lindau syndrome, multiple endocrine neoplasia type 2, neurofibromatosis type 1, and familial paraganglioma syndrome associated with mutations in the succinate dehydrogenase genes.5,6
Advances in genetic research have shown that approximately one third of these tumors are hereditary.6,7 About 10% of patients have a family history of the disease, but up to 30% of those with apparently sporadic forms have mutations inherited from their parents.6,7 The need of a genetic study in these patients is increasingly advocated, as it allows better monitoring and earlier diagnosis and treatment for patients and their relatives.6,7
Paragangliomas of extra-adrenal location are most commonly located in the head and neck, chest, abdominal, and pelvic regions. These tumors can be differentiated as functioning or non-functioning, depending on their ability or inability to secrete catecholamines, respectively. Parasympathetic paragangliomas are usually located in the head and neck and do not secrete catecholamines, manifesting essentially by mass effect. In contrast, sympathetic paragangliomas are found in abdominal and pelvic locations and usually secret catecholamines.1,2,5
The most common symptoms arise from excessive catecholamine production and usually consist of hypertension, headache, and tachycardia, among others. However, diagnosis may also be incidental, following imaging examination or family screening performed for certain syndromes.1,5
Clinical case
A 14-year-old female adolescent with unremarkable medical history was referred to the Pediatric consultation due to episodes of holocranial headache associated with blurred vision with six months of evolution. In this context, a cranial magnetic resonance imaging (MRI) was performed, with no abnormal findings.
On physical examination, stage 1 hypertension was detected and confirmed with 24-hour ambulatory blood pressure monitoring, without nocturnal dipping. No other findings were evident, such as tachycardia, diaphoresis, visual changes, nausea, vomiting, constipation, diarrhea, or weight loss. The remaining physical examination was normal and body mass index (BMI) was below the 85th percentile, according to World Health Organization curves.
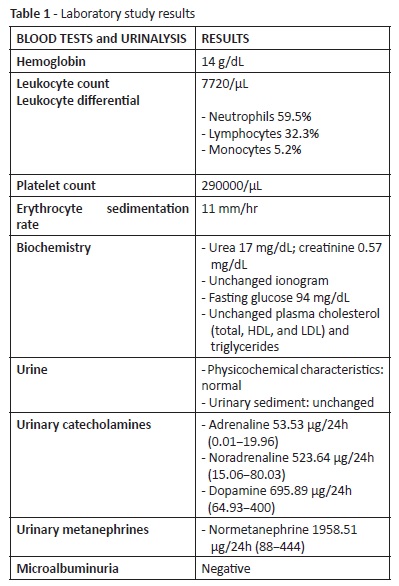
Table 1 shows laboratory assessment results. A chest X-ray, electrocardiogram, and echocardiogram were additionally performed and revealed normal.
Renal ultrasound revealed a 5.6-cm heterogeneous mass contiguous to the left renal sinus, with cystic and necrotic areas and internal vascularisation.
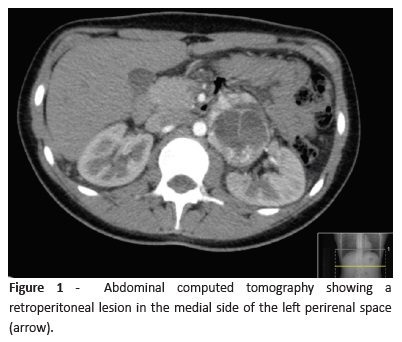
Figure 1 (abdominal computed tomography [CT]) shows a retroperitoneal lesion in the medial side of the left perirenal space, totally exophytic, multiloculated, with irregular wall and parietal thickening areas with fine and gross septations, with a 5.5-cm maximum diameter.
Anamnesis revealed that the patient’s mother had been submitted to surgery for a jugulo-tympanic paraganglioma.
Paraganglioma diagnosis relies both on tumor evidence on imaging analysis and elevated catecholamine values.
Due to retroperitoneal paraganglioma suspicion, the patient was referred to Porto’s Portuguese Institute of Oncology, where she underwent surgical excision of the lesion. Histological analysis confirmed diagnosis of sympathetic paraganglioma.
After surgery, the patient remained stable, asymptomatic, with normal blood pressure values without antihypertensive medication. Catecholamine and metanephrine values remained within the normal range, without imaging evidence of tumor recurrence.
Discussion and conclusion
Despite often underdiagnosed, hypertension in pediatric age has an estimated prevalence of 1 to 3%.
Primary or essential hypertension results from a combination of genetic and environmental factors. Incidence of the condition has increased in recent years, especially in adolescents, associated with an increase in obesity prevalence in the population.8-10
With this clinical case, the authors intend to alert to the importance of excluding a secondary cause of hypertension, particularly in cases with BMI below the 85th percentile according to World Health curves.
As noted, the major sign of these tumors is an excessive catecholamine (noradrenaline, adrenaline, and dopamine) release. They are mainly characterized by sustained or paroxysmal hypertension, headache, tachycardia, and diaphoresis. Although less frequent, nausea, vomiting, constipation, diarrhea, visual changes, and weight loss may also be present. Besides the effect of released hormones, mass effect can also be a major manifestation.2,5,7,11
Once clinical diagnosis is suspected, biochemical and imaging studies should be promptly initiated.1,3
Diagnosis relies on demonstration of excessive catecholamine levels, through measurement of serum and urinary catecholamines (adrenaline, noradrenaline, and dopamine), serum and urinary metanephrine (metanephrine and normetanephrine), and urinary vanillylmandelic acid.3,4
Measuring fractionated metanephrines (the catecholamine metabolites) in urine or plasma is the most sensitive method, as these tumors do not always continuously release catecholamines into the bloodstream and metanephrines are continuously released regardless of their precursors’ rate of secretion.4,5 Importantly, false positives may occur upon ingestion of certain foods and drugs and with physical activity.1
After biochemical study, an imaging study should be performed. This should also be performed for families at risk of pheochromocytomas and/or paragangliomas, regardless of laboratory test results.1,3 Despite its low sensitivity, ultrasound can be the initial exam performed, although CT and MRI are the preferred first choices due to their high sensitivity. MRI represents a better option for locating extra adrenal tumors.1,3,5 It is prudent to avoid using contrast in these patients, as it can trigger a hypertensive crisis.5
To complement the study, less sensitive but more specific functional imaging modalities can be employed, including I-metaiodobenzylguanidine (MIBG) scintigraphy and functional positron emission tomography (PET).3-5
After diagnosis, several authors advocate a genetic study due to the condition’s strong association with familial syndromes. In the presently reported clinical case, genetic study was key as the patient´s mother had been submitted to surgery for jugulo-tympanic paraganglioma. Syndromes associated with these tumors carry the risk of other tumors and are associated with multiple, recurrent, and sometimes malignant forms of those. Thus, genetic study has the potential to enable a more adequate follow-up, earlier diagnosis and treatment, and improved prognosis for these patients.6,7,12
The treatment of choice consists of surgical resection, achieving complete remission in around 90% of cases.1,5 Approximately two weeks before surgery, preoperative medical preparation is recommended to offset catecholamine effects, as certain stimuli, as surgery, can elicit hyperstimulation of catecholamine secretion, leading to life-threatening complications. Although consensus is lacking regarding the best option, treatment is usually performed with phenoxybenzamine, a nonselective irreversible alpha-adrenergic antagonist.5,7
Although paragangliomas are rare tumors, only accounting for approximately 1% of secondary hypertension events, this case report highlights the importance of a systematic investigation of hypertension cases in pediatric age.
REFERENCES
1. Gorostidi AM, Ranera AJ, Denis SEZ, Temprano NG, Uranga MSI, Garicano JM. Feocromocitoma y paraganglioma en la infancia: a propósito de 2 casos. An Pediatr (Barc). 2015; 82: e175-80. [ Links ]
2. Santos RJ, Domingues R, Montalvão P, Magalhães M, Bugalho MJ. Paragangliomas, caracterização clínica e funcional de 40 casos. Revista Portuguesa de Endocrinologia, Diabetes e Metabolismo 2011; 02:7-14. [ Links ]
3. Lenders JWM, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SKG, Murad MH, et al. Pheochromocytoma and Paraganglioma: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2014; 99: 1915-42. [ Links ]
4. Oliveira MC, Silva G, Machado R, Lima O, Ramires R, Marcelo F. Paraganglioma Retro-Peritoneal: Um diagnóstico raro mas importante. Acta Urológica 2009, 26; 4:47-53. [ Links ]
5. Edmonds S, Fein D, Gurtman A. Pheochromocytoma. Paediatrics in Review 2011; 32:308-10. [ Links ]
6. Ferreira MA, Vilaverde J. A genética dos feocromocitomas e paragangliomas. Rev Port Endocrinol Diabetes Metab.2014; 9:29-35. [ Links ]
7. Oleaga A, Goñi F. Feocromocitoma: actualización diagnóstica y terapêutica. Endocrinol Nutr. 2008; 55:202-16. [ Links ]
8. Correia AJM. Abordagem da Criança e Adolescentes Hipertensos. Nascer e Crescer 2007; 16:158-67. [ Links ]
9. Lurbe E, Agabiti-Rosei E, Cruickshank JK, Dominiczak A, Erdine S, Hirth A, et al. 2016 European Society of Hypertension guidelines for the management of high blood pressure in children and adolescents. Journal of Hypertension 2016; 34:1887-920. [ Links ]
10. Brady TM. Hypertension. Paediatrics in Review 2012; 33:541- 52. [ Links ]
11. Pham TH, Moir C, Thompson GB, Zarroug AE, Hamner CE, Farley D, et al. Pheochromocytoma and Paraganglioma in Children: A Review of Medical and Surgical Management at Tertiary Care Center. Paediatrics 2006; 118:1109-17. [ Links ]
12. Megías MC, Puyol DR, Rodríguez LF, Martinez GLS, Miguel PM. Feocromocitoma-paraganglioma: del diagnóstico bioquímico al genético. Nefrologia 2016; 36:481-8. [ Links ]
Endereço para correspondência | Dirección para correspondencia | Correspondence
Mariana Branco
Department of Pediatrics
Hospital de Santa Luzia
Unidade Local de Saúde do Alto Minho
Estrada de Santa Luzia 50
4901-858 Viana do Castelo
Email: mariana.a.branco@hotmail.com
Received for publication: 10.04.2018
Accepted in revised form: 07.02.2019

