










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkNascer e Crescer
versión impresa ISSN 0872-0754versión On-line ISSN 2183-9417
Nascer e Crescer vol.29 no.1 Porto ene. 2020
https://doi.org/10.25753/BirthGrowthMJ.v29.i1.18021
WHAT IS YOUR DIAGNOSIS? | QUAL O SEU DIAGNÓSTICO?
Imaging clinical case
Caso clínico imagiológico
Ana Rita BatistaI, Catarina ValpaçosI, Pedro SousaI, Teresa CostaI, Conceição MotaI, Armando ReisII, Maria Sameiro FariaI
I. Unit of Pediatric Nephrology, Department of Pediatrics, Centro Materno-Infantil do Norte. Centro Hospitalar Universitário do Porto. 4099-001 Porto, Portugal. ritagbatista@gmail.com; catarinavalpacos@gmail.com; pedromaneirasousa@gmail.com; teresa.v.c.tavares@gmail.com; conceicaocmota@gmail.com; mariasameirofaria@gmail.com
II. Unit of Pediatric Urology, Centro Hospitalar Universitário do Porto. 4099-001 Porto, Portugal. armandobreis@hotmail.com
Endereço para correspondência | Dirección para correspondencia | Correspondence
ABSTRACT
Here in is reported the case of a 16-year-old female diagnosed with vitreous haemorrhage and hemangioblastoma of the retina, referred to the Emergency Department due to sudden vision loss. Brain and pelvic magnetic resonance imaging showed cerebellar hemangioblastomas and renal nodular lesions of suspicious nature. The patient was submitted to partial left nephrectomy and histological examination revealed papillary renal cell carcinoma with clear-cell predominance. Clinical diagnosis of Von Hippel-Lindau (VHL) disease was confirmed by genetic study.
VHL disease is a hereditary, autosomal dominant syndrome of multiple neoplasms caused by germline mutations in VHL tumor-suppressor gene. Patients are predisposed to development of cysts and hypervascular neoplasms, the most common being hemangioblastomas of the central nervous system (CNS) and retina, cysts and renal cell carcinomas, and pheochromocytomas. VHL diagnosis should be suspected if an individual with family history of VHL presents with a characteristic disease lesion or, in absence of family history of VHL, with two CNS and/or retinal hemangioblastomas or a CNS/retinal hemangioblastoma associated with renal cell carcinoma, pheochromocytoma, pancreatic cysts or endocrine tumor, or epididymal cystadenoma. In VHL disease, imaging plays a key role in detection of abnormalities, follow-up, and screening of asymptomatic mutated gene carriers.
Keywords: Hemangioblastoma of the central nervous system; renal cell carcinoma hemovitreous; Von Hippel-Lindau
RESUMO
É descrito o caso clínico de uma adolescente de 16 anos diagnosticada com hemovítreo e hemangioblastoma da retina em contexto de urgência por perda súbita de visão. O estudo por ressonância magnética encefálica e pélvica evidenciou a presença de hemangioblastomas cerebelosos e lesões nodulares renais de natureza neoplásica suspeita. A doente foi submetida a nefrectomia parcial do rim esquerdo e o exame histológico confirmou o diagnóstico de carcinoma papilar de células renais com predomínio de células claras. O diagnóstico clínico de doença de Von Hippel-Lindau (VHL) foi confirmado por estudo genético.
A doença de VHL é uma síndrome hereditária autossómica dominante de neoplasias múltiplas causada por mutações germinativas no gene supressor tumoral VHL. Os doentes apresentam predisposição para o desenvolvimento de quistos e neoplasias hipervascularizadas, sendo as mais comuns hemangioblastomas do sistema nervoso central (SNC) e da retina, quistos e carcinomas de células renais e feocromocitomas. A suspeita de diagnóstico de doença de VHL deve ser considerada se um indivíduo com antecedentes familiares de VHL apresentar uma lesão característica da doença ou, em ausência de história familiar de VHL, dois hemangioblastomas do SNC e/ou retina ou um hemangioblastoma do SNC ou da retina associado a carcinoma de células renais, feocromocitoma, quistos ou tumor endócrino pancreáticos ou cistadenoma do epidídimo. A imagiologia tem um papel importante nesta condição, através do diagnóstico de anomalias, seguimento e rastreio de portadores assintomáticos.
Palavras-chave: Hemangioblastoma do sistema nervoso central; carcinoma de células renais; hemovítreo; Von Hippel-Lindau
A 16-year-old, previously healthy female with unremarkable family history (including regarding tumoral pathology) was referred to the Ophthalmology Emergency Room due to sudden unilateral loss of left vision and was diagnosed with vitreous haemorrhage and hemangioblastoma of the retina in the upper temporal quadrant. No other signs or symptoms were evident. Vision was recovered after laser photocoagulation therapy.
During follow-up in the Ophthalmology consultation, the girl underwent brain and pelvic magnetic resonance imaging (MRI) to detect other lesions.
Brain MRI (Figure 1) showed several cerebellar hemangioblastomas spread through both cerebellar hemispheres (the largest with 7-mm diameter located in the left posterior inferior medial region) associated with left ocular wall thickening and a lesion in the right supraclavicular region, probably corresponding to paraganglioma. In pelvic MRI (Figure 2), two cortical cysts (with 19-mm and 9-mm diameter) were detected in the right kidney, as well as a well-circumscribed, nodular, 9-mm lesion with low signal intensity in all sequences and several non-specific micronodules of suspected neoplastic nature in the left kidney.
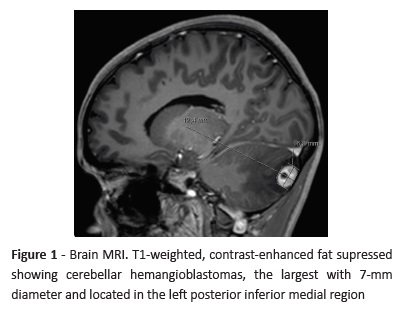
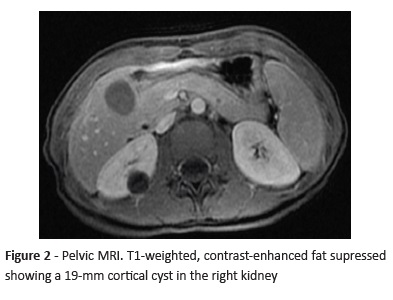
After Pediatric Nephrology/Urology consultation, the study was complemented with a reno-pelvic computed tomography (CT) scan that revealed two cortical cysts in the right kidney and several solid nodular images in the left kidney (Figura 3). Plasma as well as urinary metanephrines and cortisol laboratory results were unaltered.
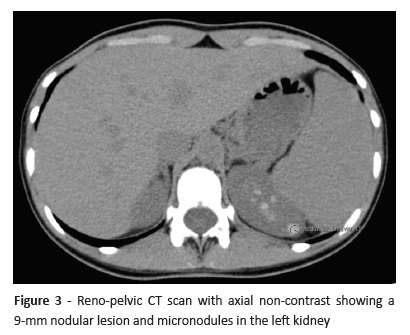
Based on imaging results, decision was made to perform a partial left nephrectomy to remove the largest left kidney lesion. Histological characterization revealed papillary renal cell carcinoma with clear-cell predominance.
The patient is currently also followed at the Neurosurgery consultation, remaining in surveillance and multidisciplinary follow-up including genetic evaluation.
What is your diagnosis?
Diagnosis
Von Hippel-Lindau disease
Discussion
Von Hippel-Lindau (VHL) disease is a rare, inherited, autosomal dominant syndrome of multiple neoplasms caused by germline mutations in the VHL tumor-suppressor gene.1,2
VHL diagnosis should be suspected when an individual with family history of VHL presents a disease-characteristic lesion, such as central nervous system (CNS) or retinal hemangioblastoma, renal cell carcinoma, pheochromocytoma, pancreatic cysts or endocrine tumors, or epididymal cystadenoma. In absence of family history of VHL, clinical diagnosis requires evidence of two tumors: two CNS and/or retinal hemangioblastomas or a CNS or retinal hemangioblastoma associated with renal cell carcinoma, pheochromocytoma, pancreatic cysts or endocrine tumor, or cystadenoma of the epididymis.1,3-6
Imaging plays a role in VHL disease, through identification of abnormalities, follow-up, and screening of asymptomatic mutated gene carriers.
The disease can be categorized in types 1 and 2. Type 1 is characterized by retinal angiomas, CNS hemangioblastomas, renal cell carcinomas, pancreatic cysts, and neuroendocrine tumors, whereas type 2 is characterized by pheochromocytomas, retinal angiomas, and CNS hemangioblastomas.7
Identification of a germline mutation on VHL gene enables molecular diagnosis of the condition.8
Patients are predisposed to the development of cysts and hypervascular neoplasms, the most common being hemangioblastomas of the CNS and retina, cysts and renal cell carcinomas, and pheochromocytomas.1,9
This case highlights the importance of thoroughly analysing key findings in the first observation, establishing early VHL diagnosis and starting immediate treatment. Retinal angiomas or hamartomas should immediately prompt additional workup to exclude VHL disease.10 In this patient, VHL molecular diagnosis was confirmed by detection of the c.326 C> G heterozygous variant in VHL gene.
During the course of disease, individuals typically manifest synchronous or metachronous neoplasms, located or not in the same organ. Although excision of hypervascular neoplasms carries hemorrhagic risks, lesion progression can potentially elicit complications that compromise patient’s quality of life and survival. Therapeutic decision is not obvious and relies on the balance between lesion progression risk and treatment.11
The uncertain tumor location and onset in VHL disease warrants lifetime follow-up. Early lesion detection in patients and affected relatives based on genetic testing allows a more conservative therapeutic approach. Additionally, mutation identification enables to predict associated phenotype and adjust the follow-up protocol accordingly.11
REFERENCES
1. Shehata BM, Stockwell CA, Castellano-Sanchez AA, Setzer S, Schmotzer CL, Robinson H. Von Hippel-Lindau (VHL) disease: an update on the clinicopathologic and genetic aspects. Adv Anat Pathol 2008; 15:165-71. [ Links ]
2. Leung RS, Biswas SV, Duncan M, Rankin S. Imaging features of von Hippel-Lindau disease. Radiographics 2008; 28:65-79. [ Links ]
3. Nordstrom-O’Brien M, van der Luijt RB, van Rooijen E, van den Ouweland AM, Majoor-Krakauer DF, Lolkema MP, et al. Genetic analysis of von Hippel-Lindau disease. Hum Mutat 2010; 31:521-37. [ Links ]
4. Shuin T, Yamasaki I, Tamura K, Okuda H, Furihata M, Ashida S. Von Hippel-Lindau disease: molecular pathological basis, clinical criteria, genetic testing, clinical features of tumors and treatment. Jpn J Clin Oncol 2006; 36:337-43. [ Links ]
5. Butman JA, Linehan WM, Lonser RR. Neurologic manifestations of von Hippel-Lindau disease. JAMA 2008; 300:1334-42. [ Links ]
6. Gatti R, Pereira MA, Neto DG. Síndrome de Von Hippel-Lindau [Von Hippel-Lindau syndrome]. Arq Bras Endocrinol Metab 1999; 43:377-88.
7. Varshney N, Kebede AA, Owusu-Dapaah H, Lather J, Kaushik M, Bhullar JS, A review of Von Hippel-Lindau syndrome. J Kidney Cancer VHL. 2017; 4:20-9. [ Links ]
8. Eamonn R. Maher. Von Hippel-Lindau Disease. Curr Mol Med 2004; 4:833-42. [ Links ]
9. Meister M, Choyke P, Anderson C, Patel U. Radiological evaluation, management, and surveillance of renal masses in Von Hippel-Lindau disease. Clin Radiol 2009; 64:589-600. [ Links ]
10. Ruppert MD, Gavin M, Mitchell KT, Peiris AN. Ocular Manifestations of von Hippel-Lindau Disease. Cureus. 2019; 11:e5319. [ Links ]
11. Gouveia S, Ribeiro C, Paiva S, Carvalheiro M. Doença de von Hippel-Lindau: da etiopatogenia ao tratamento, Rev. Port. Endocrinol Diabetes Metab. 2012; 7:28-35. [ Links ]
Endereço para correspondência | Dirección para correspondencia | Correspondence
Ana Rita Batista
Unit of Pediatric Nephrology
Department of Pediatrics
Centro Materno-Infantil do Norte
Centro Hospitalar Universitário do Porto
Largo Prof. Abel Salazar
4099-001 Porto
Email: ritagbatista@gmail.com
Received for publication: 04.06.2019
Accepted in revised form: 20.01.2020

