











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroduction
Landau-Kleffner syndrome (LKS), also known as acquired epileptic aphasia, was first described by Landau and Kleffner in 1957.1
LKS is defined as an epileptic encephalopathy, in which epileptic activity results in a deterioration of cognitive, sensory, and/or motor functions. According to several authors, LKS is currently considered a clinical variant or subtype of encephalopathy related to electrical status epilepticus during slow sleep (ESES). However, the International League Against Epilepsy (ILAE) considers the two conditions as separate entities.2
The most prominent LKS defining feature is acquired aphasia, whereas epileptic seizures are infrequent and not a prerequisite for diagnosis.3
The typical type of aphasia is verbal auditory agnosia consisting of failure to provide semantic significance to different sounds. Loss of receptive language is followed by or concurrent with expressive aphasia, a marked reduction in spontaneous speech. Other clinical manifestations include cognitive impairment and behavioral problems. Such impairment may be global or focal, based on location of epileptic discharges, and progressive, presumably related to increasing epileptiform activity.4
Herein the authors report the case of a previously healthy four-year-old boy with LKS and discuss the electroclinical features, treatment, and prognosis of this clinical entity based on a literature review.
Case report
A four-year-old boy was admitted to the Emergency Department with regression in language abilities with four days of evolution. His parents reported that he produced only a few words with no sentences and speech seemed more hesitant. Verbal auditory agnosia was also present, with the boy having difficulty in understanding what parents told him and answering common questions. Additionally, he forgot previously known words and relied on gestures to aid in communication with parents. Simultaneously, the family reported that the boy had episodes of consciousness suspension and eyelid myoclonia several times a day characterized by suddenly stopping activity, tilting the head backwards, and fluttering the eyelids. These episodes had a sudden onset and offset and usually lasted only a few seconds.
No family history of epileptic seizures was reported. Birth history was unremarkable and developmental milestones were normal until this event.
On admission, the patient’s vital data and anthropometry were normal. No facial dysmorphism was present. Neurological examination was unremarkable, except for language, as the boy was unable to understand verbal instructions and displayed occasional episodes of mostly imperceptible verbal language, even to his caregivers.
Faced with this clinical presentation, neurological and systemic causes were considered, including structural brain injuries, epileptic encephalopathies, infectious or autoimmune encephalitis, and metabolic or toxic diseases.
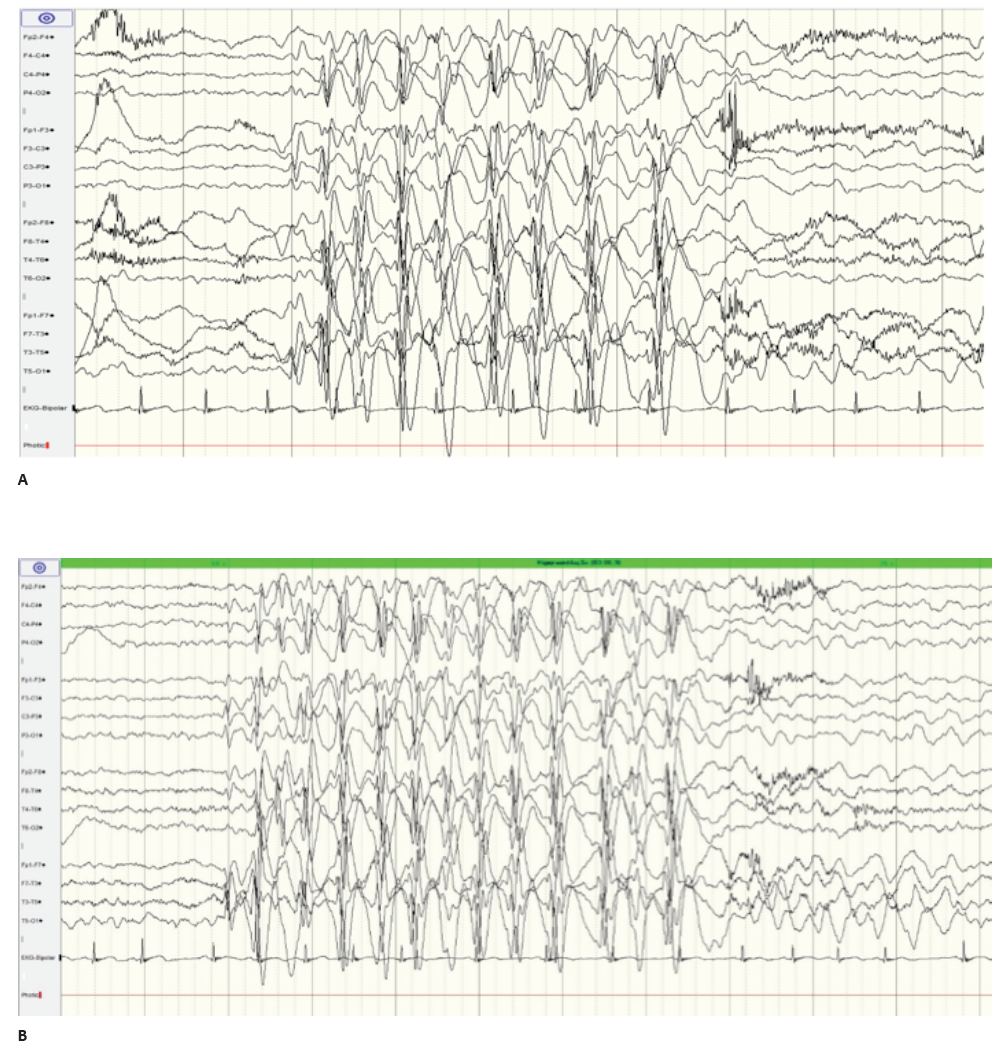
Analytical study (complete blood count, serum electrolytes, calcium, and blood glucose) was normal. Urine drug screen was negative. Brain computed tomography (CT) scan revealed no abnormality. Electroencephalogram (EEG) revealed spike-and-wave epileptiform activity prominent in the left centrotemporal region, which became virtually continuous when the child fell asleep (Figure 1and2), and generalized 2- to 3-Hertz spike-and-wave discharges, accompanied by eyelid myoclonia, occurring in wakefulness (Figure 3Aand3B). Brain magnetic resonance imaging (MRI), audiogram, and tympanogram were normal.
Once LKS diagnosis was established, treatment was started with valproate 20 mg/kg/day and clobazam 0.25 mg/kg/day for three days, followed by 0.5mg/kg/day. On the third day of treatment, seizure frequency improvement and slight verbal language recovery were observed: word output increased, although speech remained mostly imperceptible. The remaining neurological examination was normal.
Fifteen days after starting treatment, the child retrieved perception ability. He was able to understand more complex requests and answer questions easily. Marked improvement in expressive language and vocabulary was also observed, accompanied by an intelligible speech and ability to form complete sentences.
After almost three months of follow-up, ESES pattern persisted in EEG, prompting initiation of prednisolone at a dose of 4 mg/kg/day. At the end of the first month of triple therapy, EEG result returned to normal.
Clobazam was maintained for five months. High-dose corticosteroids were used for one month and tapered down over seven months. After that, the child remained on monotherapy with valproate.
When the boy completed two years of follow-up, and despite normal language function and absence of seizures, ESES pattern reappeared on EEG, prompting the addition of clobazam treatment and up-titration of valproate dosage to 28.5 mg/kg/day. Control EEG was performed two months later, showing EEG activity resolution. GRIN2A mutation screening by multiplex PCR sequencing was negative.
Currently, at the age of seven years, the boy attends the second grade of primary school with good academic performance, takes hip-hop dance classes, and shows normal neurodevelopment.
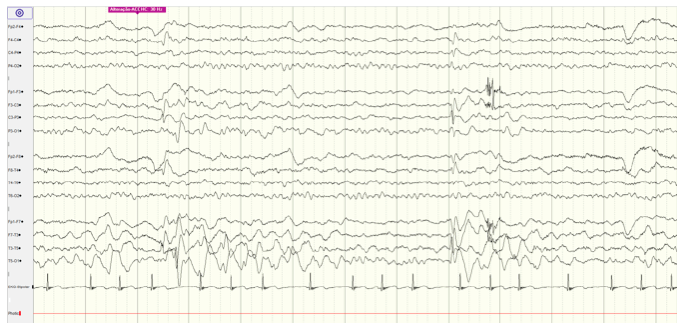
Figure 1 EEG during wakefulness showing epileptiform activity, prominent in left centrotemporal region
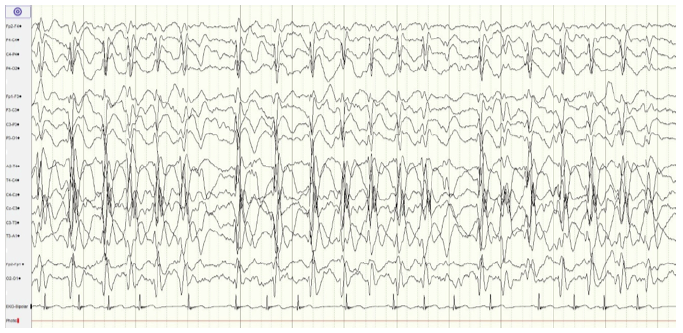
Figure 2 EEG showing spike-and-wave epileptiform activity, virtually continuous when the child fell asleep
Discussion
LKS is a rare childhood disorder with associated acquired aphasia and epileptiform EEG abnormalities.2 Although its true incidence and prevalence are unknown, boys are affected twice as frequently as girls.5 Similarly to most cases reported, in the present case the age of onset was four years.6 LKS commonly occurs between the ages of three and seven years, although onset as early as 18-22 months and as late as 13-14 years has been described.6
The disorder begins with severe disturbance of auditory language comprehension, combined with substantial expressive language disruption.6 Concordantly, the present case showed both expressive and receptive language difficulties, despite normal hearing tests.
About two thirds of children with LKS have personality disorders and behavioral disturbances. The most frequently observed behavioral problems are attentional deficits, impulsivity, distractibility, and hyperactivity.
Seizures are present in approximately 75% of cases but are usually infrequent.4 EEG abnormalities are required to establish the diagnosis.4,7-9 Generalized, bilateral, focal, or multifocal spike and wave discharges may be present, usually with central or temporal lobe predominance. When the child falls asleep, epileptiform activity becomes virtually continuous and is classified as status epilepticus during sleep (SES).4
With sophisticated imaging and nuclear medicine techniques, the epileptiform process can be shown to originate in the language cortex of the dominant temporal lobe and secondarily to spread to the homologous cortex in the other hemisphere and beyond.10 No structural abnormalities are typically seen on routine neuroimaging with CT scan or MRI. However, volumetric analysis of MRI in four children with typical LKS has shown volume reductions of 26 to 51 percent in the bilateral superior temporal areas, regions that correspond to the auditory association cortex.11,12 It is unclear whether this focal cortical atrophy is the cause of LKS or the result of intractable epileptiform activity.11,12
Language deterioration in these children may be caused by interruption of normal cortex maturation in the temporal lobes during a critical development period when the brain is making new synapses and removing others. Pervasive epileptiform activity is thought to activate and perpetuate synaptic connections that would, in the course of normal development, be removed.
The involvement of both temporal lobes eliminates the possibility that an uninvolved temporal lobe can subsume the function of the other, as often occurs in children with lesions that disrupt the dominant speech cortex early in development.4
The exact etiology of this disorder is unknown, but a genetic predisposition has been postulated.4,7 Mutations in GRIN2A gene have been described in up to 20% of individuals with LKS.4 A number of additional genes, including SRPX2,13RELN, BSN, EPHB2, and NID2,14 have also been implicated. Mutations in RBFOX1, RBFOX3, and CNKSR215 genes have been reported in patients with disorders along the epilepsy-aphasia spectrum.
Other reported etiologies include neurocysticercosis, progressive encephalitis, acute disseminated encephalomyelitis, toxoplasmosis, temporal lobe tumors, cortical malformations (eg, polymicrogyria), and vascular insults.4,7
LKS is defined as an epileptic encephalopathy. This means that the epileptic activity per se may contribute to language decline and behavioral impairment, either partially or completely.2
Therefore, this is one of the few settings in which treating EEG abnormalities, even when seizures are fully controlled or absent, may be important, as electrical epileptic activity itself is assumed to contribute to progressive cerebral function disturbance. Thus, treatment goal is not simply to control seizures, but also to eliminate the underlying EEG abnormality, in order to prevent/reverse the loss of neurologic function. Given these treatment goals, it should come as no surprise that conventional antiepileptic drugs (AEDs) are of limited benefit when used as sole therapy. If epileptic activity and language impairment persist with AEDs in the course of a few weeks, steroid therapy should be started.16 This has been done in the present case, aggressively treating the patient after diagnosis with a combination of valproate, benzodiazepines, and oral steroids, enabling, not only to suppress clinical seizures, but also to eliminate the underlying EEG abnormality to modify the course of the disease.
There is no international consensus regarding treatment of this condition. Rapid initiation of drug therapy has proven to be important for prognosis. Valproate, levetiracetam, and clobazam (or other benzodiazepines) are the most frequently used antiepileptic drugs, but can sometimes have partial or transient effects on the clinical and electrographic picture. Steroids have shown efficacy in improving both EEG activity and language.17
Intravenous immunoglobulin therapy has yielded varying results. It may nevertheless be considered in patients who are refractory to antiepileptic drugs or steroids, or in those with language impairment recurrence upon steroid withdrawal.18
Some patients may benefit from surgical treatment, but the approach must be individualized.4,19
LKS long-term outcome is not completely clear. The disease outcome is variable, and serious language disturbances occasionally remain until adulthood.20
Some studies suggest that aphasia onset before or after the age of five years has an important impact on long-term outcomes.8In the present case, it is possible that onset at the age of four years may be related to worse outcome and explain the relapse of EEG abnormalities two years after diagnosis, despite language and seizure improvement. As a result, we believe that long-term follow-up is necessary.
With this case, the authors intend to increase awareness of this rare condition, emphasizing that in one-quarter of cases clinical seizures are not present and language regression is the main characteristic feature. Early diagnosis, before global deterioration develops, appears to be crucial for effective treatment with minimal neuropsychological sequelae.
Conclusion
Landau-Kleffner syndrome is a rare, age-related encephalopathy. It occurs in previously healthy children with normal language skills and development. The exact etiology is unknown. EEG abnormalities are required for diagnosis. Early treatment is associated with improved seizure control and cognitive outcomes

