











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
Introduction
Genetic disorders presenting in the neonatal period are chronic conditions with a significant impact on the lives of patients and their families. Their clinical presentation is heterogeneous, and early diagnosis can facilitate clinical management and provide timely prognostic counseling to families. Over the past few decades, the availability of diagnostic tests has expanded, making it difficult for neonatologists (and other non-geneticists) to keep up with this progress. Appropriate use and understanding of the expanding array of genetic testing modalities require ongoing education. Herein, the authors present an overview of genetic tests currently used in the Neonatal Intensive Care Unit (NICU) and provide background on their characteristics and use in clinical practice, from classical cytogenetics to the evolving role of next-generation sequencing (NGS).
Copy number variants and single nucleotide variants
Changes in the human genome can be broadly divided into two groups: copy number variants (CNVs) and single nucleotide variants (SNVs). A CNV corresponds to a loss (deletion) or gain (e.g., duplication) of genetic information, usually greater than 50 base pairs (bp).1)-(6 On the other hand, a SNV refers to a point change in the genome.1),(5 For example, if we think of the genome as a set of 46 instruction manuals, searching for CNVs is like looking for extra or missing chapters, while searching for SNVs is like looking for changes in the letters of each word. Similarly, SNVs can be categorized as deletions, insertions, or substitutions. Considering the functional impact of SNVs on protein translation, they can be further classified as follows:5),(7
Base pair substitutions:
Silent: a base pair exchange does not imply a change in amino acid translation (given the redundant nature of the genetic code). However, not all silent changes are benign (e.g., if they occur near exon-intron transitions, they can generate alternative splicing sites).
Missense: the most common change, consisting of a base pair exchange that results in a variation in the amino acid sequence.
Nonsense: a base pair exchange that results in a premature STOP codon.
Small insertions/deletions (less than 50 bp): when an addition or subtraction of base pairs occurs, respectively. If both occur simultaneously, it is called an indel (insertion + deletion). These insertions/deletions can be further classified as follows:
In-frame: when the reading frame is maintained, i.e., the amount of deoxyribonucleic acid (DNA) lost or gained is divisible by three.
Out-of-frame: when the reading frame is altered for all subsequent nucleotides.
Frameshift: insertion or deletion of nucleotide bases in numbers that are not multiples of three, usually forming a STOP codon downstream.
Splice site: all variants that affect exon-intron transitions, thereby altering the transcribed genetic information.
Considering their relationship with the patient’s clinical status, both CNVs and SNVs can be classified into five classes based on their likelihood of pathogenicity:4), (8)-(11
Class 1: Benign (B).
Class 2: Likely benign (LB).
Class 3: Uncertain clinical relevance (Uncertain significance) (VUS).
Class 4: Likely pathogenic (LP).
Class 5: Pathogenic (P).
By convention, only the P and LP variants and VUS should be reported in laboratory reports, while the B and LB variants are usually not included.8 The classification of variants is based on available data and may therefore change over time (which is why the clinical relevance of previously identified VUS should be periodically reassessed).10),(12
In 2015, the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) published the updated standards and guidelines for the clinical interpretation of SNVs. There are 28 criteria in the ACMG/AMP guidelines, classified by weight and type of evidence. Depending on the weight of evidence, pathogenic criteria are classified as very strong (PVS1), strong (PS1-4), moderate (PM1-6), and supportive (PP1-5), while benign criteria are classified as stand-alone (BA1), strong (BS1-4), and supportive (BP1-6). These criteria are then combined according to the scoring rules to translate into the five-tier system (P/LP/VUS/LB/B). (10
However, because there is a great deal of variability in the application of these criteria, the ACMG has recently proposed a quantitative approach to reduce the classification discrepancies between different laboratories.4),(10),(11 In the case of CNVs, a score metric has been developed that assigns points to each observed piece of evidence supporting or refuting pathogenicity. CNVs with a final score ≥0.99 are considered P, while scores between 0.90 and 0.98 are considered LP. VUS are the broadest category, corresponding to scores between -0.89 and 0.89, while refuting evidence with scores between -0.90 and -0.98 or ≤-0.99 are considered LB and B, respectively.4 For SNVs, a Bayesian approach to combining rules has been incorporated, allowing the calculation of a posterior probability (Post_P). The ranges of these Post_P allow variants to be classified as P with Post_P >0.99; LP with Post_P between 0.90 and 0.99; LB with Post_P between 0.001 and 0.10; B with Post_P <0.001; and VUS with Post_P in the remainder interval.11
The next sections focus on the techniques that can be used to detect point and/or copy number variants.
Cytogenetics
Cell culture
Cell cultures are grown in a favorable artificial environment from cells obtained from an animal or plant tissue. These cultures are used as models to study cell physiology and biochemistry, as well as the effects of drugs and toxic compounds on cells.13 In human genetics, cell cultures are used to analyze tissues for clinical and/or genetic diagnosis and are an essential step in the preparation of a karyotype.9),(14 Cells can be obtained from peripheral blood leukocytes, bone marrow, amniotic fluid, or other solid tissues. Typically, the goal of these cultures is chromosome condensation and subsequent cell synchronization. Various drugs have been used for this purpose, such as methotrexate (for cell arrest in S-phase) or colchicine (to block mitotic spindle formation).14
Karyotype
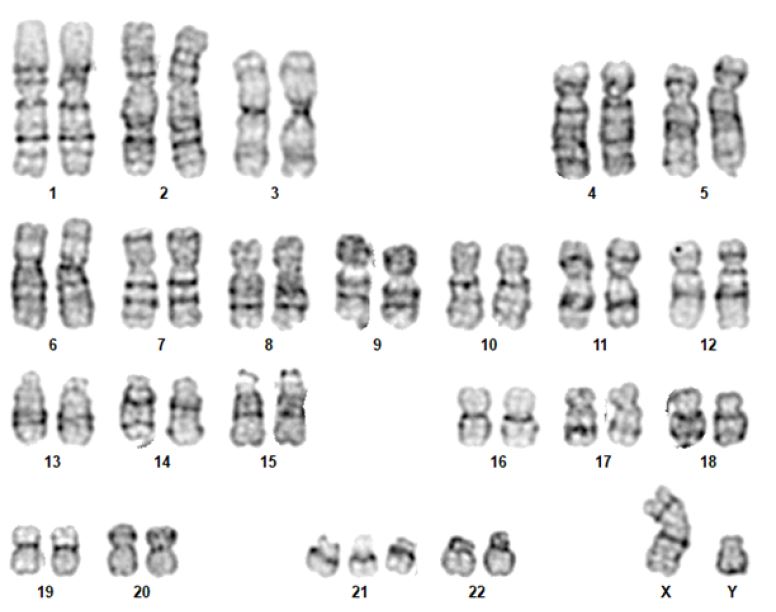
In the human species, the karyotype of a diploid cell consists of 46 chromosomes - 22 pairs of autosomes and a 23rd pair of sex chromosomes (XX, female or XY, male). Chromosomes are identified by their size, centromere position, and banding pattern.5),(14
Although the karyotype is very useful, its use as a first-line genetic test is rapidly decreasing, being currently indicated for aneuploidies (such as trisomy 13, trisomy 18, trisomy 21, monosomy X [Turner syndrome]), sexual differentiation abnormalities, and balanced chromosomal rearrangements (Figure 1).5),(8 The most commonly used staining technique is G-banding. Other staining techniques, such as Q, C (constitutional heterochromatin), R (reverse), or NOR bands (satellite chromosomes and acrocentric portions of chromosomes), can also be used. (5),(8),(14
Currently, all karyotypes are obtained with high resolution bands. This is possible by staining the chromosomes before they reach their maximum condensed state (i.e., when they are still in prophase or prometaphase). In this way, the chromosomes are more stretched, allowing a greater number of bands to be evaluated and smaller losses or gains of genetic material to be detected.5
Fluorescence in situ hybridization
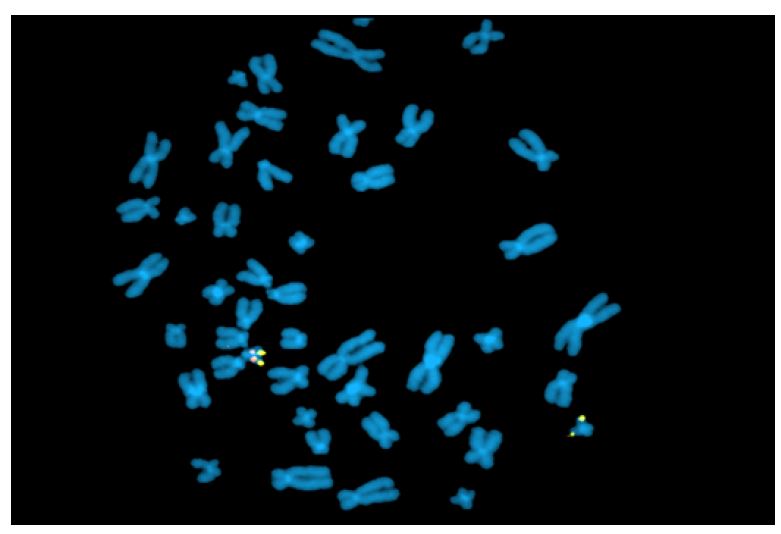
Fluorescence in situ hybridization (FISH) is a rapid analysis that uses fluorescent probes that bind to chromosomal regions with a high degree of complementarity.(5,) (12 (Figure 2) This technique must be targeted (i.e., clinicians must first specify which chromosomal region they wish to evaluate), which is different from karyotyping, where all chromosomal regions are assessed. However, both techniques are currently being abandoned in favor of other molecular techniques. Nevertheless, FISH remains one of the best methods for detecting low-grade mosaicism and is useful for describing balanced rearrangements and subtelomeric cryptic rearrangements, as well as for studying satellite chromosomes, which are not detected by comparative genomic hybridization and may also not be detected by karyotyping.3),(8
Molecular biology
Comparative genomic hybridization
Oligonucleotide array comparative genomic hybridization (oligo-array CGH or simply array CGH) allows global analysis of all chromosomes by searching for CNVs.9),(14 Array CGH is the first-line test in some clinical settings, such as intellectual disability, autism spectrum disorders, or polymalformative syndromes, and is useful in the diagnosis of conditions such as 22q11.2 microdeletions (DiGeorge syndrome), 7q11.23 microdeletions (Williams syndrome), and 1p36 microdeletions.2),(5),(8),(9
This test may also be required in other contexts, such as clarification of karyotype findings (marker chromosomes, apparently balanced translocations with an associated phenotype, breakpoint characterization) or prenatal screening of high-risk pregnancies (echographic abnormalities, intrauterine growth restriction, cardiac malformations, increased nuchal translucency).8
As a comparative technique, array CGH involves hybridizing the DNA of a control patient (of the same sex, stained red) with the DNA of the case being studied (stained green).2),(5),(12 On the plate where both DNAs are placed for hybridization, there are multiple wells (each containing a probe for a specific region of the genome).2 In situations where there is a deletion, the well will be red. If there is a duplication, the well turns green. Clinical interpretation of the results is performed using appropriate software and consulting international databases (DGV, OMIM, PubMed, DECIPHER, ClinGen), taking into account the clinical data provided by the clinician (Figure 3). (4),(9
The resolution of array CGH depends on the number of probes included. In the case of 4x180K array CGH, a total of 180,000 probes are distributed across the genome at average intervals of 13Kbp. In the case of 8x60K array CGH, the 60,000 probes are distributed at 41Kbp intervals.2),(5 On the other hand, higher resolution arrays (such as 750K array CGH) tend to include probes for both CNVs and SNPs (see below). The combination of oligo/SNP arrays has gained notoriety for its ability to simultaneously identify CNVs and loss-of-heterozygosity-associated regions (i.e., chromosomal regions that are very similar to each other because they are derived from the same ancestor).9 Typically, low-resolution array CGH is used in the prenatal context, while high-resolution array CGH is used postnatally.2),(5
In turn, single-nucleotide polymorphism genotyping arrays (SNP arrays) determine an individual’s genotype at specific polymorphic sites (i.e., loci where there are 2 or more alleles with a population frequency greater than 1%).9 The great advantage of this array technique is that it uncovers regions of homozygosity by comparing the profiles of different polymorphisms at specific chromosomal regions. This makes it possible to detect cases of parental consanguinity, uniparental isodisomy, or copy number neutral changes.9),(15
Over time, because of its higher diagnostic yield (15−20%), array CGH has largely replaced the role once played by the karyotype (~3% yield).9 This has been supported by the ability of array CGH to detect changes as small as 100 Kbp versus deletions up to 3−5 Mb or duplications greater than 5 Mb in the case of karyotype.2),(8),(9 However, array CGH has some technical limitations, such as the inability to detect balanced rearrangements, low-grade mosaicism, and supernumerary chromosomes composed of heterochromatin.3),(5),(8),(9
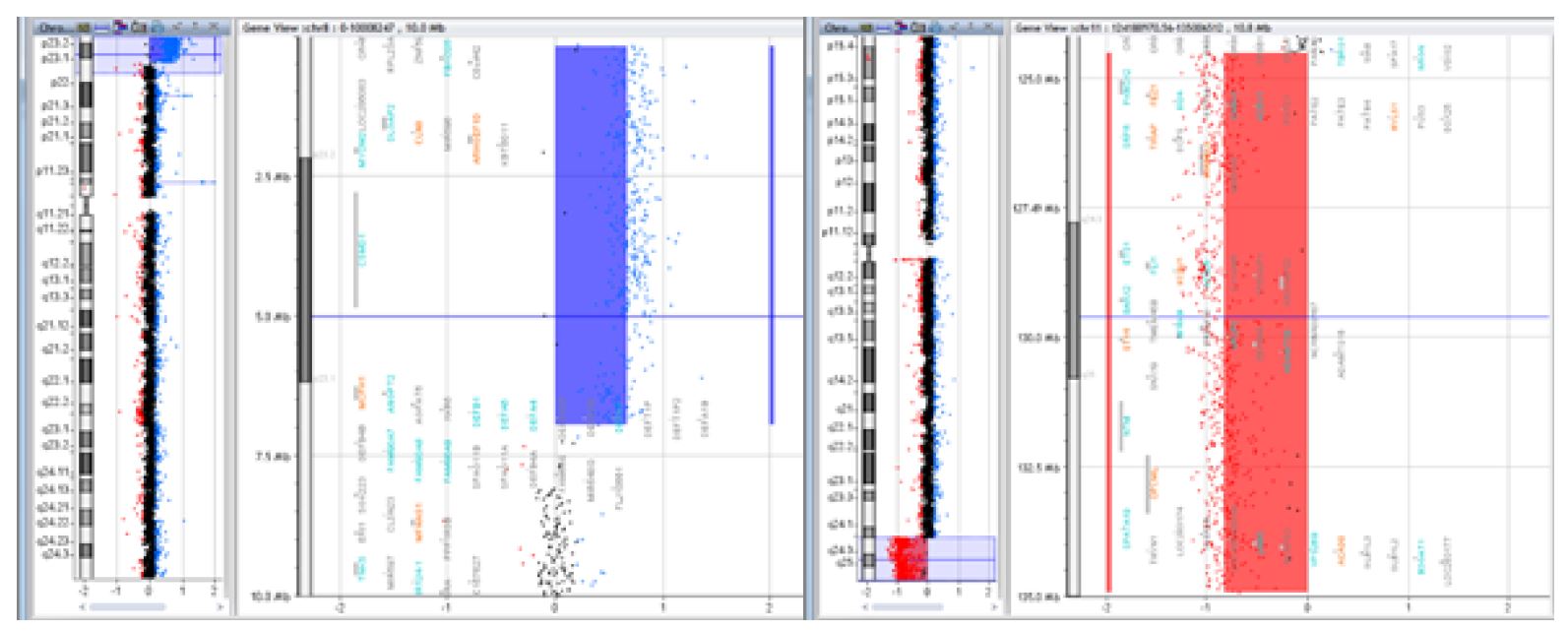
Figure 3 Case of a 15-year-old female patient with intellectual deficit, dysmorphic appearance, short stature, and normal karyotype, whose array CGH revealed a 6.7Mb deletion of the 8p23.1 region and a 10.4Mb duplication of the 11q24.2 region [arr 8p23.1-pter(176,814-6,939,296)x3, 11q24.2-qter(124,518,113-134,927,114)x1] resulting from a balanced translocation in the father
Polymerase chain reaction
One of the fundamental steps in any molecular biology protocol is polymerase chain reaction (PCR), which allows to target and amplify a genomic region of interest.5 PCR was first developed by Kary Mullis in the mid-1980s.16),(17 The following components are required to perform a PCR: two oligonucleotide primers flanking the DNA strand (both a forward, which attaches to the DNA in the 5’-3’ direction, and a reverse primer, which attaches to the DNA in the opposite direction), thermostable polymerase, nucleotides, buffer (to create an optimal pH), and water.5),(17),(18
The PCR reaction can be repeated several times, with each cycle consisting of three critical steps:5),(16),(17),(19
Denaturation (~95°C): Separation of the DNA strands by melting the double-stranded templates apart. Melting temperature depends on template length and sequence, as well as melting time.
Annealing (55-60°C): Attachment of primers targeting specific DNA sequences. Temperature depends on the primers used. Protocols often use a temperature a few degrees below the melting temperature to allow the formation of stable complexes with the target sequences and to avoid binding to other sequences.
Elongation (72ºC): Synthesis of new DNA strands using DNA polymerase to anneal the primers while stabilizing the complex.
Experimental conditions are therefore critical to the outcome of the PCR reaction. For example, if the annealing temperature is too high, there may be insufficient primer-template hybridization. If it is too low, non-specific products may be generated.17 Another point to consider is that although the PCR reaction allows for semi-quantitative evaluation, it does not allow for precise quantification of the amplified DNA. For this purpose, a quantitative PCR (qPCR) reaction can be performed. Also known as real-time PCR, qPCR was developed in 1992 and works by measuring fluorescent dyes or probes.17),(18 This allows a proportional comparison of the amount of product generated by amplification and the number of cycles required to generate a threshold amount of amplified DNA. qPCR is particularly useful in cases where there is a known CNV and the need to look for it in other family members.3),(8
Another PCR variant is triplet repeat primed PCR (TP-PCR). TP-PCR is used to detect expanded alleles in diseases whose pathological mechanism depends on triplet expansion (such as fragile X syndrome [FRAXA] or spinocerebellar ataxia). TP-PCR is particularly useful when the conventional PCR reaction identifies only one allele size (as in homoallelism, where both alleles have the same number of triplet repeats) or when allele loss is suspected (as in larger expansions).20),(21 Special probes, called triplet-primed PCR, allow the generation of fragments with a size amplitude proportional to the number of triplets that make up the expansion. The different fragments are then run on a capillary electrophoresis device, which will show a decremental pattern in the case of expanded alleles.21 If there is an interest in quantifying the expansion, long-range PCR or Southern blotting should be performed.22
Multiplex ligation-dependent probe amplification
Multiplex ligation-dependent probe amplification (MLPA) is a type of multiplex assay that quantifies the change in DNA copy number by hybridizing a probe to a target sequence.8),(12),(23 Currently, this molecular technique is the first-line test for the diagnosis of Duchene/Becker muscular dystrophy and suspected cases of spinal muscular atrophy (SMA).3
MLPA probes consist of two adjacent halves, one that binds to the 5’ region and another that binds to the 3’ region of the target sequence. In a first step, the target DNA is denatured, with subsequent hybridization of the two adjacent halves to the target region. After joining the two halves, they are combined into a single larger sequence. The resulting probe is amplified by PCR reaction. The copy number of each genomic region can be determined by comparing the amount of DNA amplified from the target region with the amount amplified from a standard sample (Figure 4).3),(12 As a comparative method, MLPA is highly susceptible to technical errors. False negative results can also occur if there are polymorphisms in the probe binding sites.3
A variation of this method, methylation-specific MLPA (MS-MLPA), is used to study epigenetic/methylation defects.3),(8),(24 DNA methylation abnormalities can result in inappropriate expression or silencing of genes.24 This test is used in disorders associated with methylation abnormalities, such as Beckwith-Wiedemann, Prader-Willi, Angelman, and Silver-Russel syndromes.3
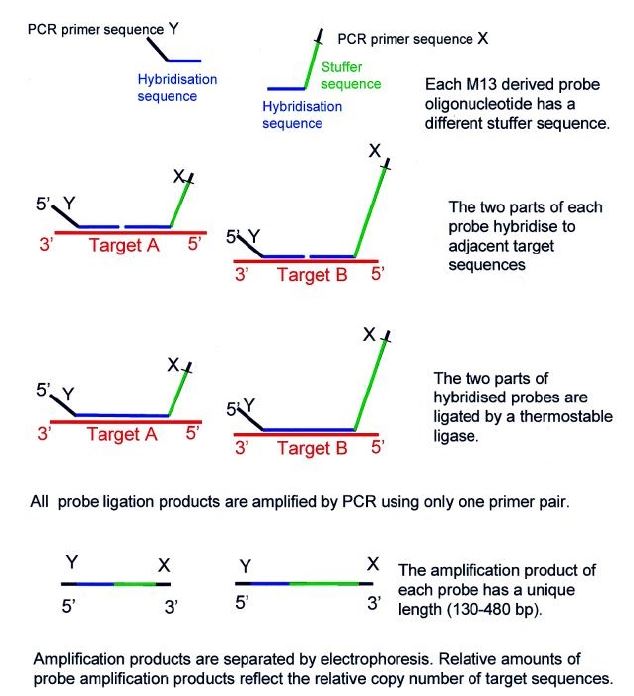
Figure 4 Multiplex ligation-dependent probe amplification (MLPA) scheme. Reprinted from Schouten JP et al.23
Sanger sequencing
In the 1970s, Frederick Sanger described the first sequencing technique, which became the basis for the Human Genome Project (HGP).25),(26 Sanger sequencing (also known as the chain termination method) uses special nucleotides called dideoxy terminators (ddNTPs), which are characterized by the absence of the free OH group at the 3’ carbon of the pentose.5),(25),(27 When these ddNTPs are added to a growing DNA sequence, the sequence is prevented from continuing to grow. This results in DNA sequences of different sizes which, after amplification, are run on an agarose gel.5),(27 Depending on the radioactivity of each fragment and its size, it is possible to determine the genetic sequence of the target region nucleotide by nucleotide.5),(25
Nowadays, instead of using radioactive ddNTPs or agarose gels, automated methods based on fluorescent ddNTPs and running fragments in multichannel capillary electrophoresis devices are used (Figure 5).25),(27 This method is still time-consuming and very expensive if the intention is to sequence a large number of genes (justifying the years and billions of dollars spent on the HGP to sequence a single genome).12),(26 However, this does not invalidate its value, with this method currently considered not only the most reliable, but also the gold standard (often used as a confirmatory test for NGS). In addition, Sanger sequencing can be used for family studies, especially when a pathogenic variant has been identified in the affected proband. (10),(11
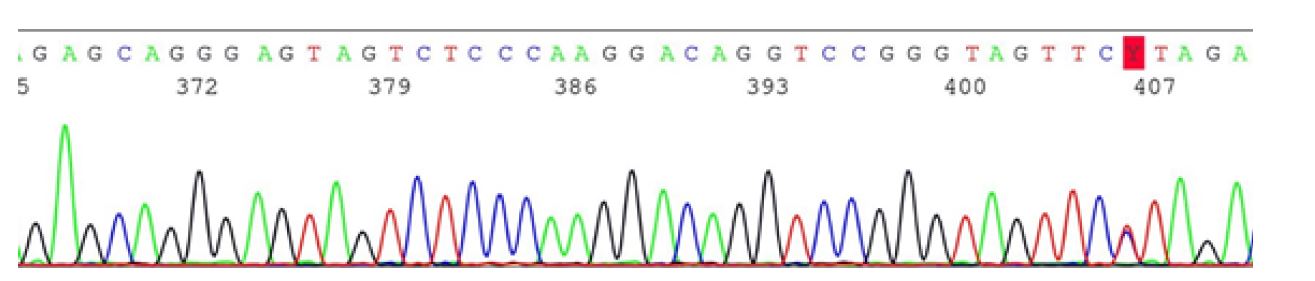
Next-generation sequencing
NGS, or high-throughput nucleotide sequencing, is based on the parallel sequencing of different genomic sequences. This is done by fragmenting the genome into smaller sequences (about 100-200 bp) that are amplified simultaneously (short reads, collectively called a DNA library). The subsequent alignment of the different fragments into a single sequence allows the detection of variants in an exon or exon-intron transition (by mapping the DNA short reads to the reference genome).5),(25 Thus, the main advantage of NGS is its ease of use and speed in obtaining results, since it can sequence the entire human genome in a single day (with the associated time and cost expected to decrease in the coming times).26),(28
Today, a larger number of second-generation NGS platforms are available. Overall, second-generation methods can be divided into two major groups: sequencing by hybridization and sequencing by synthesis.25),(27 Most commercially available sequence by synthesis platforms use reversible nucleotides that can reverse the temporary block of DNA strand growth (unlikely ddNTPs). Of these, the following can be highlighted:25),(27),(28
Ion Torrent: Also known as ion exchange sequencing. A hydrogen ion is released upon incorporation of a new nucleotide, which changes the medium pH. This voltage change is detected by a pH sensor, which instantly converts the sequencing phenomenon into digital information, speeding up the process.
Illumina technology: The most widely used method of sequencing by synthesis due to its greater ease of use. It is based on the concept of bridged PCR using fluorescent nucleotides.29
One of the major limitations of NGS is its inability to sequence large fragments. While Sanger sequencing can read sequences from 600 to >1000 bp, NGS methods can only read shorter sequences (from 300 to 500 bp).25),(27 In this sense, third- and fourth-generation methods (such as PacBio or Nanopore) have been developed in recent years in an effort to combine the advantages of NGS - sequencing multiple fragments simultaneously - with those of Sanger - sequencing larger fragments, reducing the probability of reading errors.25),(28),(30),(31
Because NGS allows multiple genes to be analyzed simultaneously, they are grouped into gene panels for clinical convenience:5),(8),(9
Gene-specific panel: assesses all genes associated with a given phenotype, from only a few to hundreds of genes.
Mendeliome or clinical exome: assesses all gene exons associated with OMIM phenotypes (which represent approximately 1-2% of the entire human genome and include approximately 85% of all variants described to date).
Whole exome sequencing (WES): assesses all gene exons, even those of unknown function. Useful for non-specific phenotypes and when all other tests are negative.
Whole genome sequencing (WGS): assesses the entire genome and may in the future replace the role currently played by array CGH and WES. WGS is currently used in clinical research and has little applicability in clinical practice.
Both WES and WGS can be performed alone or, for example, in trio (simultaneously comparing the genome of a child and its parents).5),(10),(32 The rationale for this strategy is to reduce the amount of useless information generated by sequencing methods (for example, if the VUS identified are inherited, they are less likely to be disease-causing because a healthy parent is also a carrier).5 This is because the larger the panel examined, the lower its specificity and the greater the number of VUS identified.9),(12 The large amount of data generated by NGS, as well as its difficult interpretation, causes serious and complex problems that overload not only the workflow of laboratory pipelines, but also the medical team, which spends a lot of time discussing and trying to assign meaning to these variants.33 Other limitations of NGS should also be considered, such as the inability to detect balanced rearrangements, nucleotide expansions, uniparental disomy, or methylation defects.8),(9),(33
Critical considerations about genetic testing
When ordering a genetic test, the pros and cons should always be weighed. To avoid misdiagnosis, a targeted diagnostic test (e.g., Sanger sequencing, FISH, MLPA, or gene panels) should be ordered when a specific syndrome is suspected.32),(34 Advantages include better coverage of the gene(s) of interest and less chance of equivocal results.9),(12 Examples of targeted diagnostic tests include MLPA for SMA, FBN1 gene sequencing for Marfan syndrome, or multigene panels for Noonan syndrome.5),(12
However, an untargeted test may be useful if the clinical presentation is nonspecific (e.g., karyotype, array CGH, WES, or WGS). When performing some of these more comprehensive studies (especially WES or WGS), one should consider the possibility of incidental findings that may reveal pathogenic variants unrelated to the phenotype (e.g., predisposition to cancer or neurodegeneration). Carrier status should also be considered.33),(35 These findings should be given the greatest relevance. Patients should receive genetic counseling throughout the process and their autonomy should be respected.4),(5),(12 In some countries, such as Portugal, genetic counseling is mandatory before genetic testing is ordered for certain conditions.36 In the case of minors, when genetic testing is recommended for diagnostic purposes, parents must consent to the results they wish to receive after discussing the benefits and risks.5),(36
The best interests of the child should not be disregarded. When appropriate, the child should be given the opportunity to express his/her opinion on the request for genetic studies, and his/her will should not be devalued, whatever it may be.37 The American Academy of Paediatrics (AAP) and the ACMG emphasize that predictive genetic testing is inappropriate for asymptomatic minors at risk for adult-onset diseases for which early treatment has no beneficial effect, because it denies the child’s autonomy.5),(12),(37),(38 The same concerns are shared by the Portuguese law.36
Another aspect to be considered is the confidentiality of genetic data. Several countries have anti-discrimination laws both in access to health insurance and in employment. This means that everyone can know their genetic data and make important lifestyle and medical decisions without fear of genetic discrimination.39),(40
Conclusions
Technological advances in the field of genetics over the past few decades have opened up previously unimagined possibilities. In clinical practice, comparative genomic hybridization has largely replaced the role once played by the karyotype in detecting unbalanced rearrangements. NGS has made an unprecedented contribution to the identification of new monogenetic causes of disease, which would have taken years of collaborative work using Sanger sequencing alone. However, it is not enough to simply read the genome. It is necessary to interpret it and understand whether certain individual variants are simply different versions of the normal or disease-causing. Thus, collaboration between different specialties and fields of knowledge is essential in the arduous mission of decoding the human genome.
Acknowledgements
The authors gratefully acknowledge the collaboration of Prof. Sofia Dória and Dr. Susana Ferreira, from the Genetics Service of the Faculty of Medicine of the University of Porto (FMUP), for providing the figures that complement the text of this article.
Authorship
Diogo Fernandes da Rocha - Investigation; Methodology; Validation; Visualization; Writing - original draft
Gustavo Rocha - Conceptualization; Supervision; Validation; Writing - review & editing
Pedro Louro - Supervision; Validation; Writing - review & editing
Glossary
Acrocentric chromosomes: chromosomes in which the centromere is not in the middle but near the end. Humans usually have five pairs of acrocentric autosomes (chromosomes 13, 14, 15, 21, 22). The Y chromosome is also acrocentric.
Allele: one of two or more versions of a DNA sequence (a single base or a segment of bases) at a given genomic location.
Allele dropout: event that occurs when there is a polymorphism in the site where the primers should bind, resulting in poor or no amplification of one or both alleles for a given individual.
Aneuploidy: abnormal number of chromosomes in a cell that does not include a difference of one or more complete sets of chromosomes (e.g., having 45 or 47 chromosomes).
Balanced rearrangement: type of chromosomal structural variant (e.g., translocation, inversion, or insertion) without apparent cytogenetic gain or loss of chromatin.
Cell synchronization: process by which cells at different stages of the cell cycle are brought to the same phase.
Codon: nucleotide triplets that correspond to amino acids or stop signals during translation.
Constitutional heterochromatin: chromatin thought to maintain a condensed and transcriptionally inert chromatin conformation (formed primarily at gene-poor pericentromeric regions).
Copy-neutral change: phenomenon whereby one of two homologous chromosomal regions is lost, but various mechanisms have ensured the presence of two identical copies of such a region in the genome, thus making it copy neutral.
Marker chromosomes: structurally abnormal, extra pieces of unidentified chromosomal material that usually occur in addition to the normal chromosomal complement. Also referred to as supernumerary chromosomes.
Mosaicism: a condition in which the same person has two or more sets of cells that are genetically different. If 20−40% of cells are abnormal, it is considered low-grade mosaicism.
Nucleotide expansion: diseases associated with nucleotide expansions occur when the number of triplets present in a mutated gene is greater than the number found in a normal gene. These are called dynamic mutations.
OMIM: catalog of all known human genes and genetic phenotypes, freely available and updated daily. The official home is omim.org.
Reading frame: the division of a sequence of nucleotides into a set of consecutive, non-overlapping triplets.
Satellite chromosome: the end of a chromosome that is separated from the rest of the chromosome by a secondary constriction. Found in acrocentric chromosomes.
Sequencing by hybridization: indirect sequencing method in which sets of oligonucleotides are hybridized under conditions that allow the detection of complementary sequences in the target nucleic acid.
Sequencing by synthesis: polymerase-dependent sequencing approach in which the sequencing reaction generates a newly synthesized DNA strand.
Subtelomeric cryptic rearrangement: chromosomal rearrangement involving the end of chromosomes (telomeres).
Supernumerary chromosome: the same as Marker chromosome.
Thermostable polymerase: a special type of polymerase that can withstand the higher temperatures used in polymerase chain reaction (PCR) while maintaining enzymatic activity.
Uniparental disomy: when the two copies of a chromosome come from the same parent, rather than one from the mother and one from the father. In uniparental isodisomy, the two copies come from one chromosome, while in uniparental heterodisomy, the two copies come from each chromosome.

