











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
Introduction
Dent disease (DD) is a rare X-linked renal proximal tubular disorder characterized by low molecular weight (LMW) proteinuria and hypercalciuria. Nephrocalcinosis and nephrolithiasis may also be present, leading over time to chronic kidney disease (CKD).1-8
DD comprises two genetic subtypes: up to 60% of patients have inactivating pathogenic variants of the CLCN5 gene at chromosome Xp11.23 (Dent-1), and about 15% have pathogenic OCRL variants at chromosome Xq26.1 encoding a phosphatidylinositol 4,5-biphosphate 5-phosphatase (Dent-2).1,2,4 No specific gene defect has been described in the remaining 25%.2,4
The renal phenotype of Dent-2 is comparable to that of Dent-1, except for a lower prevalence of nephrocalcinosis.1,3,5 Since CLCN5 and OCRL1 mutations impair the megalin-cubilin system (MCS), loss of LMW proteins is a universal finding in DD, and persistent proteinuria is a known renal injury.1,4,5 Hypercalciuria is present in up to 90% of patients with confirmed DD.5) Other signs of proximal tubule dysfunction, namely glycosuria, aminoaciduria, and phosphaturia, are also common features, and mild hypophosphatemia occurs in approximately one third of patients.1,4,5
The disease has a marked male predominance, with symptomatic disease mainly confined to males.1-6 In the first decade of life, children may manifest only LMW proteinuria and/or hypercalciuria, both of which are usually asymptomatic, making the diagnosis quite challenging.1,3,4
Herein are reported three cases of DD referred to a Pediatric Nephrology Department for asymptomatic proteinuria first detected during screening for an acute condition (complementary data on Table 1).
Table 1 Clinical and anthropometric data of male patients with Dent disease at the time of initial evaluation and complementary examination. For laboratory parameters, the results shown correspond to the time of diagnosis.
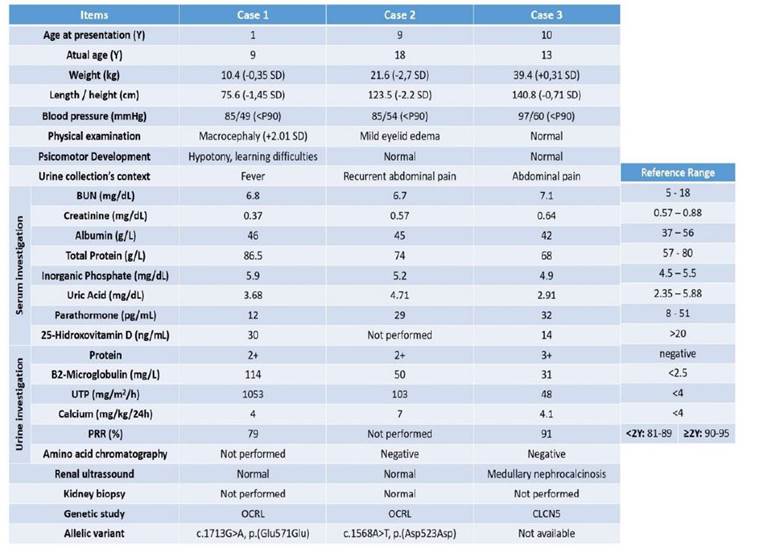
BUN - Blood Urea Nitrogen; P- Percentile; PRR - Phosphorus reabsortion rate; SD - Standard deviation, UTP - Urinary total protein; Y - Years
Clinical cases
Case 1:
A fifteen-month-old male child was diagnosed with proteinuria during an episode of acute febrile tonsillitis. His personal and family history was unremarkable (table 1). His daily milk intake was approximately 500 milliliters and he was on vitamin D supplementation. The boy had macrocephaly (50.3 cm; +2.01 standard deviation [SD]) with ventriculomegaly. Mild hypotonia was noted initially, and learning difficulties were noted later in the preschool years. The remaining physical examination was unremarkable. Laboratory studies revealed mixed (tubular and glomerular) nephrotic proteinuria (NP) and intermittent hypercalciuria. DNA sequencing revealed a pathogenic mutation in the maternally inherited OCRL gene.
The last analytical control at the age of nine years (without treatment) showed normal kidney function and a protein/creatinine ratio in occasional urine sample of 1.97 mg/mg.
Case 2:
A nine-year-old boy presented with proteinuria detected on urinalysis requested due to recurrent abdominal pain. He had an acute febrile nasopharyngitis the previous week (table 1). The boy played football twice a week, but there was no history of trauma and no apparent temporal relationship of the condition to exercise, and no relevant family or personal history except for low weight and height for age. The parents reported predominantly morning eyelid edema. There were no other significant findings on examination.
Laboratory study revealed non-orthostatic mixed NP and hypercalciuria. Renal ultrasound and blood pressure were normal. At ten years of age, the boy was treated with enalapril, with incremental daily doses up to 0.3 mg/kg because of the increase in urea and creatinine. Three years later, height-weight deceleration was evident and subsequent endocrine evaluation was negative. Kidney biopsy was normal and genetic study revealed a missense hemizygous variant in the OCRL gene.
At the time of the last observation, at the age of eighteen years, before the transition to adult care, the boy had no complaints and there were no physical findings. The last analytical evaluation, under enalapril, showed normal kidney function and a protein/creatinine ratio in occasional urine sample of 1.80 mg/mg.
Case 3:
A ten-year-old boy presented with proteinuria during evaluation for abdominal pain associated with Campylobacter jejuni gastroenteritis. His personal and family history was unremarkable (table 1).
Physical examination revealed no abnormalities except for pectus excavatum. Complementary study revealed non-orthostatic mixed NP and intermittent hypercalciuria. Renal ultrasound was compatible with medullary nephrocalcinosis. DNA sequencing revealed a hemizygous pathogenic deletion in the CLCN5 gene.
At 13 years of age, the boy’s renal function was normal and he maintained significant non-nephrotic proteinuria (36 mg/m2/hour) without treatment.
Discussion
DD is a progressive kidney disease characterized by hypercalciuria, LMW proteinuria, and other signs of proximal tubular dysfunction such as hypophosphatemia and acidosis.1-8 Clinical diagnosis requires evidence of LMW proteinuria, hypercalciuria, and at least one of the following criteria: nephrocalcinosis, nephrolithiasis, hypophosphatemia, hematuria, or renal insufficiency.1-8 However, in children under the age of ten, only LMW proteinuria and/or hypercalciuria may be present, both of which are usually asymptomatic.1-8) Molecular testing confirms the diagnosis.
The three reported cases (table 1) share several characteristics: they were all males who presented with asymptomatic non-orthostatic NP detected by urinalysis in the setting of screening for an acute condition. According to the literature, half of patients with DD have proteinuria in the nephrotic range, which is often present in early childhood and typically increases with age.1,4,5 Therefore, despite the mixed NP due to MCS impairment, these patients usually have normal serum albumin levels and do not develop nephrotic syndrome.1,4-6
Although always detected, hypercalciuria was intermittent, which may have delayed the diagnosis of DD.1,8 Moreover, hypercalciuria may even resolve completely with dietary calcium restriction, suggesting intestinal hyperabsorption as the main pathophysiologic mechanism.1,3,5,6
In addition to these renal disease features suggesting selective proximal tubulopathy rather than full Fanconi syndrome, a mutation found in the CLCN5 (case 3) and OCRL (cases 1 and 2) genes confirmed DD.
Furthermore, no alterations in glomerular filtration, lipid profile, autoimmunity, or phosphocalcium metabolism (including hypophosphatemia) have been observed in any of the cases to date. Although approximately 25% of affected males may develop rickets or osteomalacia, most children have normal bone mineral density and attain normal height.1,5,6 As illustrated in the second clinical case, patients may have growth restriction and short stature, which is typically mild and subclinical in Dent-1 and more pronounced in Dent-2.1,5,6 Children with Dent-2 may also have developmental delay and/or mild intellectual impairment, as illustrated in the first clinical case.1,5,6
Nephrocalcinosis was present in case 2 and it is thought to occur in up to 75% of cases, often in childhood.1,6,7
Kidney biopsy is an invasive diagnostic test with unspecific findings.7 It was performed in the second clinical case because of unexplained persistent NP that was not controlled with increasing doses of enalapril, was associated with height-weight deceleration, and predicted significant delay in obtaining molecular test result.
The primary goals of treatment are to reduce hypercalciuria, prevent nephrocalcinosis, and delay progression to CKD.1,2 Angiotensin-converting enzyme inhibitors (ACEI) may reduce proteinuria and delay loss of kidney function, but data on their effectiveness are sparse.1,2 ACEI (enalapril) were used only in case 2 in this study due to the persistence of NP on serial analyses.
Approximately two thirds of affected males develop some degree of CKD, with decreased creatinine clearance usually becoming apparent in late childhood.1,6 The patient in case 2 had already reached adulthood and no decline in kidney function was observed. For cases 1 and 3, both clinical and analytical follow-up will be mandatory in the coming years.
Overall, the diagnosis of DD is challenging, especially in the absence of a family history, and requires a high degree of clinical suspicion. It should be considered in all male patients with unexplained proteinuria, as it may allow earlier diagnosis and avoid unnecessary kidney biopsies and potentially harmful therapies.1,4,8)
Authorship
Inês Rosinha - Conceptualization; Data curation; Formal analysis; Writing - original draft; Writing - review & editing
Marta Machado - Conceptualization; Formal analysis
Ana Carolina Cordinhã - Conceptualization; Formal analysis
Carmen do Carmo - Conceptualization; Formal analysis
Clara Gomes - Conceptualization; Formal analysis

