










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortugaliae Electrochimica Acta
versão impressa ISSN 0872-1904
Port. Electrochim. Acta v.28 n.3 Coimbra 2010
Role of Halides on the Passivation of Iron in Alkaline Buffer Solutions
S. Nathira Begum, C. Ahmed Basha,* V.S. Muralidharan
Central Electrochemical Research Institute, Karaikudi-630 006, India
DOI: 10.4152/pea.201003143
Abstract
Cyclic voltammetric studies were carried out on pure iron in alkaline borate and phosphate buffer solutions at pH 10.8. At higher potentials, on anodic polarization, iron forms FeB4O7 and FeOOH in borate buffer, and FeHPO4 in phosphate buffer which got converted to higher valency phosphates.
In phosphate solutions, in presence of halides, the interfacial diffusion layer turned to be cation selective outer sublayer and an anion selective inner sublayer, and in borate solutions a precipitate layer of metal oxyhydroxide was formed, which was anion selective, and anions adsorb on this.
Keywords: iron, cyclic voltammetry, passivity, alkaline corrosion, interfaces, anodic films.
Introduction
Over a long period it was supposed that the electrons are the only particles taking part in metal dissolution [1]. When metal dissolves anions and solvent molecules participate in complex formation, the adsorbed water forms surface charge transfer complexes (SCTC) with surface atoms of the transition metals. Studies on iron dissolution in acetonitrile solutions with small amounts of water confirmed this [2]. The formation of adsorbed complexes [M-H2O]ads is accompanied by a partial charge transfer from the water molecules to the metal. In presence of various anions, iron dissolution was explained invoking the participation of SCTC and mechanistic schemes.
Many studies about active dissolution and passivation were carried in alkaline solution [3-12] and many electrochemical studies have been investigating the behaviour of iron in aqueous solution. Several mechanisms have been suggested [3-9] but no consensus has yet been found by different authors. However, the majority of authors propose the existence of an intermediate transient species, Fe(I), generally designated as FeOHads, although there is little, if any, direct evidence for its existence [4,13-14]. In the presence of a sufficient quantity of OH- and / or an aggressive anion, A-, competitive adsorption between water molecules and anions may occur, which means that surface attack occurs by forming Fe [H2O] or Fe(A-) groups [4]. If A- forms an iron soluble salt, the corrosion is not uniform. If the salt is insoluble, A- may assist passivation depending on its capacity for homogeneously covering the metallic surface increasing the ionic charge transfer resistance [4]. The species Fe[H2O] or Fe(OH-) initiate the oxidation process by electrochemical deprotonation reactions [4, 5, 14]. Under potentiodynamic conditions, the first anodic peak is specifically related to Fe(OH)2 formation and the second peak to Fe(OH)2 with three dimensional oxide film, and the third peak to FeOOH formation [15-18]. From the variation of the peak height as a function of sweep rate and hydroxide concentration [19], the film growth on the surface was assigned to low field migration of ions through an oxide/hydroxide lattice. Again, the three anodic peaks observed [20, 21] had been assigned to the ionization of adsorbed hydrogen, Fe(OH)2 and Fe3O4 formation.
The present investigation deals with the participation of halide ions in the passivation of iron in alkaline borate and phosphate buffer solutions. Cyclic voltammetric studies were carried out to understand the influence of halides on the iron dissolution and passivation.
Materials and methods
The working electrode was made of pure iron rod (99.9999% purity, Johnson Matthey Chemicals Ltd., U.K) with a circular area of 0.2 cm2. The rod was embedded in Teflon gaskets and electrical connections were provided by screw and thread arrangement. The surface of the electrode was polished successively with finest grade emery papers and with 0.05 µm alumina, degreased with trichloroethylene and washed with running double distilled water. The counter electrode was a platinum sheet of 4 cm2 area and a Hg/HgO/OH- ion electrode was used as reference. A potentiostat /galvanostat Model IM6 was used for obtaining cyclic voltammograms (CVs). The experiments were carried out in borate and phosphate buffer solutions kept at pH 10.8 in presence and absence of different concentrations of halide ions (Cl-, Br- and I-). All solutions were deoxygenated with purified N2. Each experiment was performed with freshly prepared solutions and on freshly prepared surface. All measurements were performed at 30 ± 1 °C. The potentiodynamic polarization curves were recorded by changing the potential automatically at the desired sweep rate.
In order to start with a clean surface, the electrode was kept at -1.5 V or -1.2 V, depending on the buffer solution, for half an hour, disconnected shaken free of adsorbed hydrogen and then subjected to triangular potential scan at various sweep rates. The potential range -1.3 V (EΛc) to +1.2 V (EΛa) was fixed at a sweep rate of 100 mVs-1 after several experiments to get reproducible potential versus current.
Results and discussion
The results of the experiments carried out are presented in Figs. 1 to 5 and Table 1. Oxidation and reduction processes occurring on an iron electrode in alkali solutions were reviewed and reported [16, 17]. The first anodic peak in cyclic voltammograms is specially due to Fe(OH)2 formation, the second anodic peak to Fe(OH)2 with three dimensional oxide films, and the third peak to FeOOH formation [17,18]. Oxidation of iron occurs as
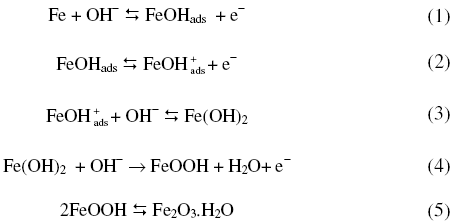
X-ray diffraction studies on oxide of iron [22] revealed that Fe2O3 to be present in all passive films independent of pH. Ellipsometric responses of iron electrode in alkali showed [23] a composite structure of the passivating layer involving an inner layer, which is difficult to electro-reduce probably related to Fe3O4, and an outer gelatinous iron hydroxide layer which is reducible. One is a compact barrier film adjacent to the metal, and another is an outer strongly hydrated film [24].
Electrochemical behaviour in phosphate buffer solutions
When polarized from -1200 mV to +100 mV, the forward scan exhibited a peak at -395 mV. On reversing the scan, zero current crossing potential (ZCCP) appeared at -404 mV. A cathodic peak appeared at -590 mV (Fig. 1). Appearance of an anodic peak at -395 mV suggests that iron was oxidized to divalent state and the oxidation proceeds in pH 10.8 as
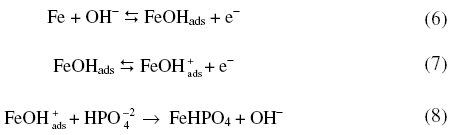
The phosphate layer thickened with increase in anodic potentials. While reversing the scan the appearance of a cathodic peak was due to
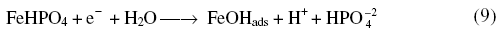
which underwent reduction to iron at extreme cathodic potentials along with H2.
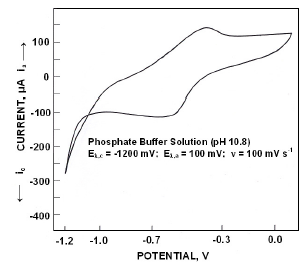
Figure 1. Cyclic voltammograms for iron in phosphate buffer solution of pH 10.8.
Fig. 2a presents the electrochemical spectrum obtained in presence of various concentrations of chloride ions. An anodic peak appeared at -410 mV whose peak potentials and peak currents did not vary with chloride concentration. On reversing the scan, a cathodic peak appeared at -450 mV. Cathodic peak potentials became noble while peak currents decreased with chloride concentration.
When polarized from -1200 mV to +100 mV, the forward scan in different concentrations of bromide ions (Fig. 2b) exhibited an anodic peak at -400 mV whose peak potentials and peak currents did not vary with concentration. Cathodic peak appeared in the reverse scan at -625 mV. Cathodic peak potentials became active while the peak currents were invariant.
Fig. 2c presents the electrochemical behaviour in different concentrations of iodide ions. An anodic peak appeared at -400 mV. Anodic peak currents and potentials were invariant with iodide concentration. On reversing the scan a cathodic peak appeared at -600 mV. Cathodic peak potentials did not vary with iodide ion concentrations while the peak currents decreased.
Role of anions on the dissolution, passivation and pitting was reviewed recently [25]. In phosphate solutions, iron phosphate along with iron hydroxyl polymers may be formed in the interfacial diffusion layer. These metal complexes are immobile and may provide a positively charged layer similar to ion selective membranes. In presence of halides, the interfacial layer gets modified. On anodic polarization, adsorption of phosphate ions on the surface takes place with oxygen atoms of the phosphate group pointing towards the metal. The outer layer surface being positively charged facilitates the adsorption of halides, consequently the interfacial diffusion layer turns to be cation selective outer sub layer originally present and an anion selective inner sublayer. Such a bipolar layer is resistive to ferrous ion transport, the reverse bipolarity to anodic current. The observed invariance of anodic and cathodic peak currents in presence of halide ion concentration confirms this.
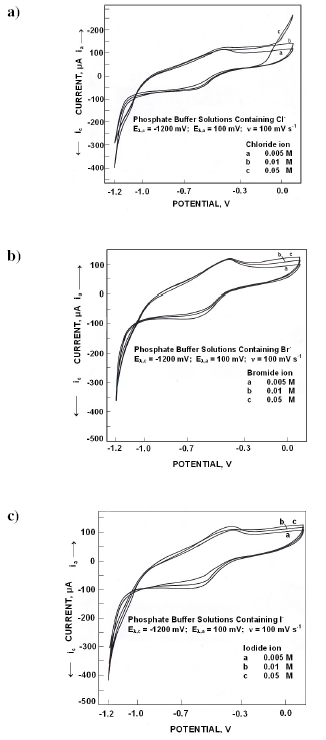
Figure 2. Cyclic voltammograms for iron in phosphate buffer solutions of pH 10.8 containing different concentrations of a) Cl- ions, b) Br- ions and c) I- ions.
Electrochemical behaviour in borate buffer solutions
Fig.3 presents the electrochemical spectrum obtained in borate buffer solutions, when polarized from -1500 mV to +500 mV the forward scan exhibited an anodic peak at -883 mV (I) followed by a broad peak in the range of -300 mV (II). On reversing the scan a cathodic peak appeared at -592 mV (III) followed by a peak (IV) at -1330 mV.
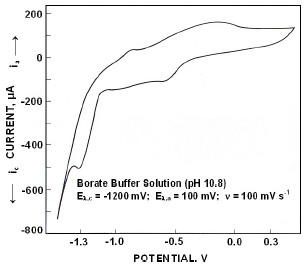
Figure 3. Cyclic voltammograms for iron in borate buffer solutions of pH 10.8.
In the presence of chloride ions the electrochemical spectrum was modified (Fig. 4a). During the forward scan in 0.005 M Cl- solutions, an anodic peak (I) appeared at -140 mV, whose peak potentials became active with increase in Cl- ion concentration; current crossing appeared at +150 mV. While reversing the scan, two cathodic peaks appeared at -700 mV (II) and at -1200 mV (III). Cathodic peak potentials became nobler with chloride ion concentration.
Fig. 4b presents the cyclic voltammograms obtained in various concentrations of bromide ions. Broad anodic peak (I) appeared at -310 mV in 0.005 M solutions which became active with increase in concentration. While reversing the scan around +150 mV, current crossing appeared. Cathodic peaks appeared at -670 mV (II) and at -1100 mV (III). Cathodic peak potentials became nobler with bromide ion concentration.
When polarized from -1500 mV to +500 mV, the forward scan exhibited a shoulder (I) at -850 mV followed by a peak at -350 mV (II) (Fig. 4c). Anodic peak potentials (II) became active with increase in iodide ion concentrations. While reversing the scan, a cathodic peak appeared at -650 mV (III) followed by a peak at -1350 mV (IV). Cathodic peak potentials became nobler with iodide ion concentration.
In borate buffer solutions, the interfacial layer consists of ferrous hydroxo complexes and their polymers. These metal complex ions and polymer ions are less mobile than simply hydrated ions. With progress in polymerization of metal hydroxo complexes, a precipitate layer of metal oxyhydroxide may be formed which is anion selective. Anions get adsorbed on the surface.
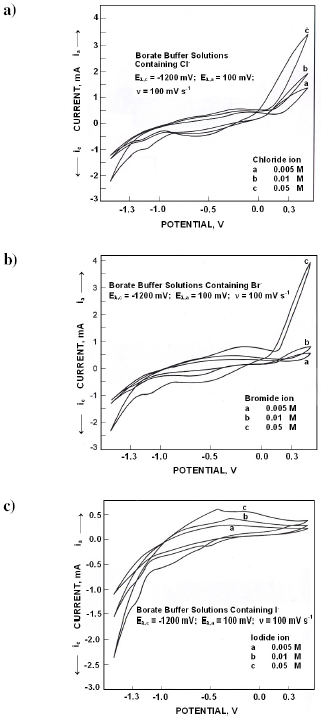
Figure 4. Cyclic voltammograms for iron in borate buffer solutions of pH 10.8 containing different concentrations of a) Cl- ions, b) Br- ions and c) I- ions.
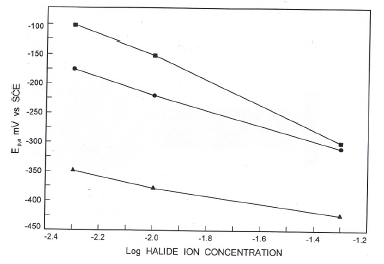
Figure 5. Variation of Ep,a with log [halide ion concentration].
Fig.5 presents the variation of anodic peak potential with log (halide ion concentration) in borate buffer solutions. The variation of Epa with log [X-] suggests the participation of halide ions
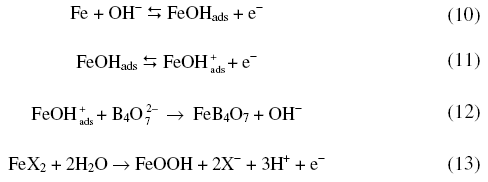
where X- is a halide ion. Increase of halide ion concentration enhanced the anodic peak currents confirming the participation of halide ions (Table 1).
Table 1. Variation of anodic peak currents with halide concentration.

Conclusions
Iron underwent passivation in alkaline buffered phosphate and borate solutions. On anodic polarization iron formed FeHPO4 which got converted to higher valent phosphates at higher potentials in phosphate solutions. In borate buffer solutions, iron forms FeB4O7 and FeOOH on higher potentials.
In phosphate solutions in presence of halides, the interfacial layer got modified. The interfacial diffusion layer turned to be cation selective outer sublayer originally present and an anion selective inner sublayer. In borate solutions a precipitate layer of metal oxyhydroxide was formed, which was anion selective and anions adsorb on this.
Acknowledgement
Our sincere thanks are due to the Director, Central Electrochemical Research Institute for all his encouragements.
References
1. B. Kabanov, R. Burstein, A. Frumkin, Disc. Faraday Soc. 259 (1974) 1. [ Links ]
2. Ya. Kolotrykin, M. Lazorenko, R.M. Manevich, L.A. Sokolova, Electrokhimiya 30 (1994) 537.
3. S.T. Amaral, I.L. Muller, Corros. Sci. 41 (1999) 759. [10.1016/S0010-938X(98)00149-8]
4. J.R. Vilche, A.J. Arvia, Anal. Acad. Nac. Cs. Ex. Fis. Nat. 33 (1981) 100.
5. D.M. Drazic, C.S. Chao, Electrochim. Acta 27 (1982) 1409. [10.1016/0013-4686(82)80031-5]
6. J. Zou, D. Chin, Electrochim. Acta 33 (1988) 477. [10.1016/0013-4686(88)80164-6]
7. J. Zou, D. Chin, Electrochim. Acta 32 (1987) 1751. [10.1016/0013-4686(87)80010-5]
8. J. Flis, H. Oranowska, Z. Szklarska-Smialowska, Corros. Sci. 30 (1990) 1085. [10.1016/0010-938X(90)90058-D]
9. R.D. Armstrong, I. Baurhoo, J. Electroanal. Chem. 34 (1972) 41. [10.1016/S0022-0728(72)80500-X]
10. D.D. MacDonald, D. Owen, J. Electrochem. Soc. 123 (1976) 824.
11. D.D. MacDonald, B. Roberts, Electrochim. Acta 23 (1978) 557. [10.1016/0013-4686(78)85036-1]
12. D.D. MacDonald, B. Roberts, Electrochim. Acta 23 (1978) 781. [10.1016/0013-4686(78)80039-5]
13. G.T. Burstein, G.M. Ashley, Corrosion 39 (1983) 241.
14. J.O. Zerbino, J.R. Vilche, A.J. Arvia, J. Appl. Electrochem. 11 (1981) 703. [10.1007/BF00615174]
15. M. Jayalakshmi, V.S. Muralidharan, Corros. Rev. 12 (1994) 305.
16. S. Nathira Begum, V.S. Muralidharan, C. Ahmed Basha, Applied Surface Science 254 (2008) 2325. [10.1016/j.apsusc.2007.09.019]
17. R.S. Schrebler-Guzman, J.R. Vilche, A.J. Arvia, Electrochim. Acta 24 (1979) 395. [10.1016/0013-4686(79)87026-7]
18. R.S. Schrebler-Guzman, J.R. Vilche, A.J. Arvia, J. Appl. Electrochem. 11 (1981) 551. [10.1007/BF00616674]
19. D. MacDonald, B. Roberts, Electrochim. Acta 23 (1978) 781. [10.1016/0013-4686(78)80039-5]
20. D. Geana, A.A. Ei Miligy, W.J. Lorenz, J. Appl. Electrochem. 4 (1974) 337. [10.1007/BF00608976]
21. G. Kreysa, B. Hakansson, Electrochim. Acta 23 (1978) 61.
22. D.W. Shoesmith, P. Taylor, M.G. Bailey, B. Ikeda, Electrochim Acta 23 (1978) 903. [10.1016/0013-4686(78)87014-5
23. O.A. Albani, J.O. Zerbino, J.R. Vilche, A.J. Arvia, Electrochim Acta 31 (1986) 1403. [10.1016/0013-4686(86)87052-9
24. N. Sato, K. Kudo, R. Nishimura, J. Electrochem. Soc. 123 (1976) 1419. [10.1149/1.2132612
25. V.S. Muralidharan, Corrosion Reviews 21-4 (2003) 327.
Received 14 November 2009; accepted 08 April 2010
* Corresponding author: basha@cecri.res.in, cab_50@rediffmail.com

