










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkPortugaliae Electrochimica Acta
versión impresa ISSN 0872-1904
Port. Electrochim. Acta vol.29 no.3 Coimbra jul. 2011
Voltammetric Reduction Behavior and Electrode Kinetics
K. Balaji*, K. Reddaiah, T.M. Reddy and S.R.J. Reddy
Electrochemical Research Laboratories, Department of Chemistry, Sri Venkateswara University, Tirupati-517502, India
Abstract
The electrochemical reduction behavior of cefepime and cefpirome has been studied using cyclic voltammetry in Britton Robinson (BR) buffer ranging from pH 2.0 to 12.0. Cefepime and cefpirome are found to give two well defined peaks and these peaks are attributed to the reduction of the azomethine group (>C=N-) by two electron process in two steps. Kinetic parameters such as diffusion coefficient (D) and forward rate constant (Kof,h) values are evaluated and a reduction mechanism is proposed. The result indicates that the process of both the compounds is irreversible and diffusion controlled. A simple and rapid differential pulse polarographic method has been developed for the determination of cefepime and cefpirome in pharmaceutical formulations and spiked urine samples using the standard addition method. The lower detection limits are found to be 4.6×10-8 mol. L-1 and 8.52×10-8 mol. L-1 for cefepime and cefpirome, respectively.
Keywords: cefepime, cefpirome, cyclic voltammetry, differential pulse polarography, pharmaceutical formulations, urine samples.
Introduction
Cephalosporins, which have been in use since 1948, are semisynthetic antibiotics of the β-lactam family, because of their convenient antibacterial activity, β-lactamase resistance, and pharmacokinetic properties [1-3]. They have the advantages of being applicable to penicillinic-allergic patients, active against penicillin resistant microorganisms in some instances, and also used for therapy of a variety of infections [1,2], developed in intensive care units. These antibiotics have assumed a prominent role in modern antimicrobial therapy due to the enhanced intrinsic microbiological activities and favorable safety profile. Cefepime and cefpirome (Fig. 1) are new parenteral cephalosporins that have been described as a fourth generation, broad-spectrum cephalosphorin [4,5]. Cefepime and cefpirome are structurally similar to other aminothiazolyl methoxyimino third-generation cephalosporins, cefotaxime and ceftazidime.
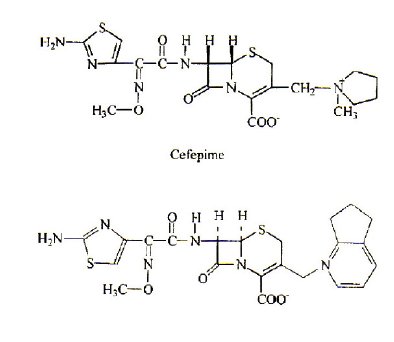
Figure 1. Cefepime
The electrochemical reactions of azomethine compounds have been one of the most active areas of research in cathodic electrochemistry. Cephalosporins contain >C=N- group, which plays an important role in the development of organic electrochemistry. Polarographic determination of cephalosporins has appeared in a number of papers [6-10]. The main electrode reaction responsible for polarographic activity of cephalosporins is found to be the reduction of azomethine group. Because of the biological and scientific values of the cephalosporins, it is important to get clear information on electrode reaction mechanistic aspects.
Different methods have been used for the determination of cefepime, i.e., high-performance liquid chromatography and simple spectrophotometric methods [5, 11-14]. However, although the selectivity and the detection limit have been improved in these methods, these are rather time consuming methods and require large number of complicated steps to follow on for analysis. Therefore, a simple technique with high sensitivity and selectivity has been desired for the detection of cefepime and cefpirome.
Polarographic and voltammetric techniques such as differential pulse polarography [6,10,15] and adsorptive stripping voltammetry [16] are employed for the determination of cephalosporins such as ceftazidime, ceftizoxime, ceftriaxone, cefazolin, cefuroxime, and cefotaxime. There are a few published papers concerning voltammetric behaviour and determination of cefepime in human fluids such as serum, urine and cerebrospinal fluid [17]. The analytical problems described here for this determination are of considerable value to analytical chemistry. The purpose of the present work is to study the voltammetric reduction behavior of cefepime and cefpirome by employing cyclic voltammetry and to establish the methodology for their trace determination by using differential pulse voltammetry in pharmaceutical formulations and in urine samples.
Experimental
Materials and reagents
Cefepime and cefpirome were purchased from Cipla labs India Ltd., (Mumbai). The samples are used directly without further purification. Standard stock solutions (1×10-3 mol L-1) are prepared by dissolving an appropriate amount of electroactive species in deionized triple distilled water. The standard stock solutions are protected from light throughout the experimental procedure.
Instrumentation
Differential Pulse Polarographic (DPP) measurements and Cyclic Voltammetric (CV) measurements were performed with Metrohm-757 VA computrace controlled by computer running electrochemical analysis software; output was Hewlett packed plotter. Three electrodes assembly consisted of a dropping mercury electrode or hanging mercury electrode as a working electrode, an Ag/AgCl (Metrohm 6.0728.000) as reference electrode, and a glassy carbon (Metrohm 6.1247.000) as auxiliary electrode. pH measurements were carried out by using a Metrohm-632 pH meter. Controlled potential electrolysis was performed with a Techno Potentiostat (Model PS-603). All measurements were carried out at room temperature. IR spectra were recorded with a Perkin-Elmer 1600 spectrometer.
Results and discussion
Characterization of waves/peaks
The voltammetric behaviour of cefepime and cefpirome has been examined in pH range 2.0 to 12.0 by using differential pulse polarography and cyclic voltammetry. Cefepime and cefpirome have been found to give two peaks in the entire buffer system and these peaks are attributed to the reduction of azomethine group by two-electron process in two steps. The cyclic voltammogram and differential pulse polarogram of cefepime are shown in Figs. 2 and 3.
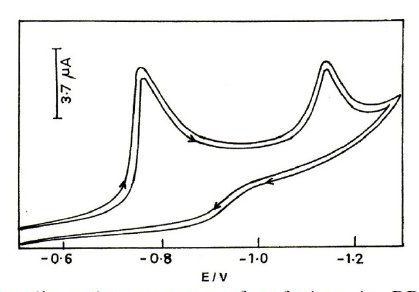
Figure 2. Typical cyclic voltammogram of cefepime in BR buffer of pH 2.0; concentration 0.5 mM; scan rate 50 mV/s.
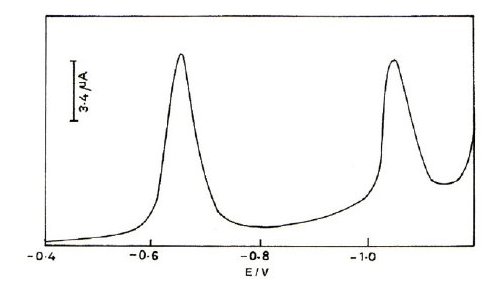
Figure 3. Typical differential pulse polarogram of cefepime in BR buffer of pH 2.0; concentration 0.5mM; pulse amplitude 50 mV; drop time 2s.
Nature of the electrode process
The reversibility of the reduction process is studied at HMDE using cyclic voltammetry. No anodic peak is observed in the pH range for the compound. The nature of the reduction process is found to be diffusion-controlled and adsorption free in the buffer system studied, as evidenced from the linear plots of im vs. t2/3 [18, 19] and ip vs. v1/2 relationships, where im is the maximum current in differential pulse polarography, t is the drop time, ip is the peak current in cyclic voltammetry, and v is the scan rate. The peak potential (Em) values of cefepime and cefpirome are dependent on pH and shifted towards more negative potentials along with an increase in the pH of the buffer system, indicating proton involvement in the electrode process. The shifting of Em values towards more negative potentials on increasing the concentration of the electroactive species, indicating the irreversibility of the electrode process. The optimum pH for high resolved peaks was obtained at pH 2.0. In the basic buffers of pH > 8, the curves are very poor due to the non-availability of protons and are not useful for analytical purpose. It is interesting to note that cefepime and cefpirome are found to be reduced even in strongly basic media. This behaviour can be attributed to the presence of methoxy group, which is directly attached to the N-atom of the >C=N- group. Hence, methoxy group will facilitate the reduction of the >C=N- group.
Identification of reduction products
Controlled potential electrolysis is carried out for the identification of reduction products. 2 mL of 1×10-3 mol L-1 solution of the electroactive species are placed in the cell and the electrolysis are carried out at a potential -1.04 V and -1.01 V vs. (Ag/AgCl) for respective compounds at pH 2.0. During the electrolysis, solutions are continuously stirred and purged with nitrogen. After the electrolysis the solutions are extracted three times with 20 mL of ethyl acetate. The combined extracts are dried over anhydrous sodium sulphate and the solvent removed by evaporation. The results indicate that the product is a saturated product and it is confirmed as an amino group (range 3250 cm-1) which is an evidence from IR spectra. Millicoulometry is employed to find out the number of electrons involved in the electrode process using the method of Devries and Kroon [20] and is found to be four for the both compounds in the entire buffer system.
Kinetic data
Tables 1 and 2 show the kinetic parameters such as transfer coefficient, diffusion coefficient and heterogeneous forward rate constant for cefepime. The diffusion coefficient values are found to be in good agreement in all the techniques, showing the adsorption free nature of the electrodes process in all the techniques.
Table 1. Typical cyclic voltammetric data of cefepime. Concentration 0.5 mM; scan rate: 50 mV s-1.
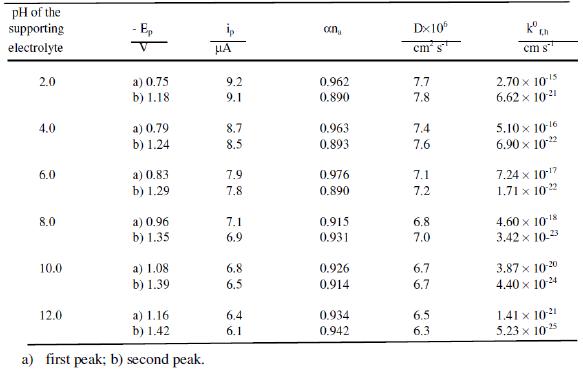
Table 2. Typical differential pulse polarographic data of cefepime. Concentration 0.5 mM; drop time: 2s.
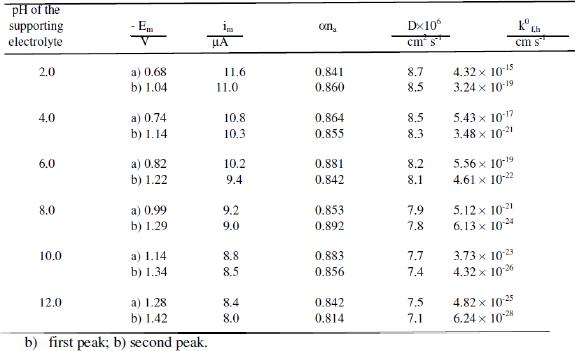
The heterogeneous forward rate constant values are found to decrease with increase of the pH of the buffer solution, which may account for the shift of reduction potentials towards more negative values with increase in pH. This trend evident in the proton transfer is involved in the electrode process. Possibly an increase in pH increases the dissociation constant of the protonated species and these factors affect the protonation rate. Consequently, the reduction potential is shifted to more negative values.
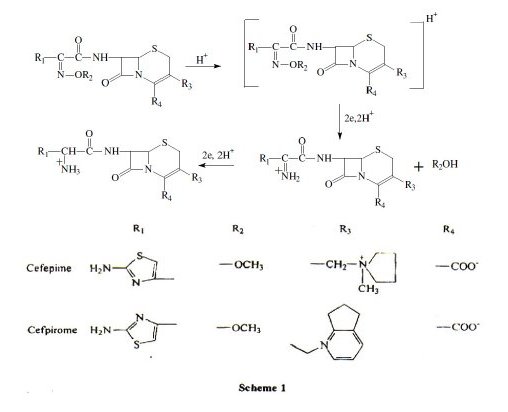
Electrode mechanism
Based on the above results, the following mechanism may be assigned for the electrochemical reduction of cefepime and cefpirome (scheme 1).
Analysis
For the purpose of analysis the optimum pH for the compounds cefepime and cefpirome is pH 2.0. The obtained first peak is sharp and reproducible; it is preferred for the analysis. For the estimation of the drugs in pharmaceutical formulations and in spiked urine samples the standard addition and calibration methods were employed. The peak current for cefepime and cefpirome is found to be linear over the range of concentration 8.5×10-8 to 1.5×10-5 mol L-1 and 6.3×10-8 to 1.3×10-5 mol L-1. The lower detection limit is found to be 8.5×10-8 mol L-1 and 4.6×10-8 mol L-1. The detection limit is calculated using the expression dl = 3 SD/m, where SD is the standard deviation and 'm' is the slope of the calibration plot.
Recommended analytical procedure
Ten milliliters of the BR buffer solution are deoxygenated in the cell with oxygen free nitrogen gas. An aliquot of standard solution of the electroactive species is added to the buffer contained in the cell. After recording the polarograms, small increments (0.2 mL) of standard solution are added and polarograms are recorded after each addition under the same conditions. The optimum conditions for cefepime and cefpirome in pH 2.0 are found to be of drop time 2 s, pulse amplitude 50 mV and applied potentials of -0.68 V, -1.04 V and -0.59 V, -1.01 V, respectively. The above described analytical procedure has been employed for the determination of these drugs in pharmaceutical formulations and urine samples.
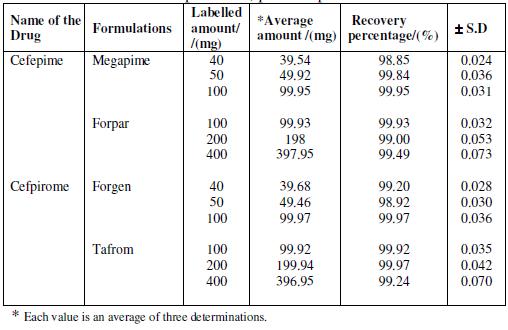
Analysis of pharmaceutical dosage forms
The proposed procedure for the determination of cefepime (Megapime-500 mg, Forpar-500 mg) and cefpirome (Forgen-500 mg, Tafrom-g) is applied to four Indian commercialized pharmaceutical formulations (injections). Four injection samples of each are fully dissolved in deionized distilled water and transferred into a 1 L calibrated flask. Known volumes (0.5 mL) of the clear supernatant liquid are diluted to 10 mL with supporting electrolyte and subjected to polarography. The amount of the compound in the injection is calculated by standard addition method. The accurate and precise results obtained for the formulation of cefepime and cefpirome are found to be in good agreement with those of the declared values of each drug taken from different formulations. The results obtained for the determination of the drugs in pharmaceutical formulations are given in Table 3.
Table 3. Differential pulse polarographic data of cefepime and cefpirome in pharmaceutical formulations. Drop time 2 s; pulse amplitude 50 mV.
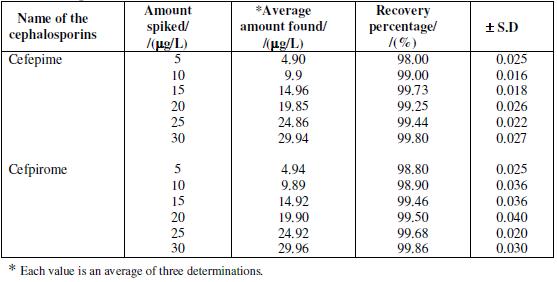
Analysis of spiked urine samples
8 h (overnight) or 24 h collected urine (fresh or refrigerated and equilibrated to room temperature) was transferred to a 500 mL flask. An appropriate amount of cefepime and cefpirome standard solution was added and the flask was made up with urine to the mark. The spiked urine was either analyzed immediately or aliquots were put in a freezer until measurements were performed. When analyzing, deep frozen samples were first left at room temperature in dark until thermally equilibrated while they were sporadically shaken. There was no interference from the metabolites and the results obtained for the determination of drug in spiked urine samples are given in Table 4.
Table 4. Determination of cefepime and cefpirome in urine samples: pulse amplitude of 50 mV; drop time of 2 s.
Section of analysis
A linear relationship is found over the concentration range 8.5×10-8 to 1.5×10-5 mol L-1 and 6.3×10-8 to 1.3×10-5 mol L-1 for cefepime and cefpirome, respectively; the limit of detection is given by the expression LOD = 3 SD/m [7], where 'SD' is the standard deviation of replicate determination values and 'm' is the slop of the calibration curve. Now SD = 1.35×10-3 and 1.42×10-3 μA, m = 8.8×104 and 5×104 μA mol L-1, hence LOD = 3×SD/m = 3×1.35×10-3 /8.8 ×104 = 4.6×10-8 mol. L-1 and 3×1.42×10-3 /5×104 = 8.52×10-8 mol L-1. The LOQ is defined as 10 SD/m [21], and is found to be 1.53×10-7 mol L-1 and 2.84×10-7 mol L-1, respectively.
Conclusion
The electrochemical reduction behaviours of cefepime and cefpirome are found to be irreversible, diffusion controlled and free from adsorption from the cyclic voltammetry and differential pulse polarography techniques. The peak potentials are observed to be pH dependent and are found to be shifted cathodically with increase in pH of the buffer system studied, indicating proton involvement in the electrode process. The heterogeneous forward rate constant value for the reduction of the cefepime and cefpirome is found to decrease with increase in pH. This shows that the electrode process tends to become more and more irreversible with increase of the pH of the solution. By employing differential pulse polarography analytical procedures are described for the quantification of the above compounds. Both standard addition and calibration methods are employed for the estimation of these pharmacologically important drugs in their pharmaceutical formulations, and urine samples.
References
1. P. Garzone, J.A. Lyon, V.L. Yu, Drug Intell. Clin. Pharm. 17 (1983) 507. [ Links ]
2. P. Garzone, J.A. Lyon, V.L. Yu, Drug Intell. Clin. Pharm. 17 (1983) 615. [ Links ]
3. B. Van Klingeren, L.J. Van Wijngaarden, A. Rutgers, J. Antimicrob. Chemother. 674 (1980) 676. [ Links ]
4. R.E. Hancock, F. Bellido, J. Chemother. 29 (1992) 1. [ Links ]
5. R.H. Barbhaiya, C.A. Knupp, S.T. Forgue, G.R. Matzke, C.E .Halstenson, J.A. Opsahl, K.A. Pittman, Drug Metab. Dispos. 19 (1991) 68. [ Links ]
6. G.V.S. Reddy, S.J. Reddy, Talanta 44 (1997) 627. [ Links ]
7. T.M. Reddy, M. Sreedhar, S.J. Reddy, J. Pharm. Biomed. Anal. 31 (2003) 811. [ Links ]
8. B. Schroder, R. Voigt, R. Patsch, G. Horn, F.B. Spenker, Fresenius Z Anal. Chem. 331 (1988) 529. [ Links ]
9. E. Munoz, L. Camacho, J. L. Avila, F. Garcia-Blanco, Analyst, 113 (1988) 23. [ Links ]
10. M.S.S. Ragavan, Y.V. Rami Reddy, S. Jayarama Reddy, J. Electrochem. Soc. India 41 (1992) 165.
11. H. Elkhaili, L. Linger, H. Monteil, F. Jehl, J. Chromatogr. B 690 (1997) 181. [ Links ]
12. S.M. Rabouan-Guyon, A.F. Guet, P.Y. Courtois, D.M.C. Barthes, Int. J. Pharm. 154 (1997) 185. [ Links ]
13. C.E. Fasching, L.R. Peterson, D.N. Gerding, J. Liq. Chromatogr. 9 (1986) 1803. [ Links ]
14. V. Rodenas, A. Parra, J. Garcia-Villanova, M.D. Gomez, J. Pharm. Biomed. Anal. 13 (1995) 1095. [ Links ]
15. M.J. Hernandez, P.A. Sanchez, Z.M. Delgado, L.L. Vega, Anal. Chim. Acta 160 (1984) 335. [ Links ]
16. B. Ogorevc, A. Krasna, V. Hudnik, S. Gomiscek, Microchim. Acta [Wien] I (1991) 131. [ Links ]
17. F.J.J. Palacios, M.C. Mochon, J.C.J. Sanchez, J.H. Carranza, J. Pharm. Sci. 92 (2003) 1854. [ Links ]
18. M.R. Smyth, J. Osteryoung, Anal. Chem. Acta 96 (1978) 335. [ Links ]
19. M. Sreedhar, J. Damodar, N.V.V. Jyothi, S.J. Reddy, Bull. Chem. Soc. Jpn.73 (2000) 2477. [ Links ]
20. T. Devries, J.L. Kroon, J. Am. Chem. Soc. 75 (1953) 2484. [ Links ]
21. B. Uslu, S.A. Ozkan, Anal. Chem. Acta 462 (2002) 49.
*Corresponding author. E-mail address: hammoutib@gmail.com
Received 27 December 2010; accepted 21 April 2011

